
Blog
A Drug for PSSM?
20 Feb 2026

INTRODUCTION
A new drug that inhibits glycogen synthase I, encoded by GYS1, reduces muscle glycogen in a mouse model of Pompe disease, a deficiency of the enzyme alpha-1,4-glucosidase, encoded by GAA (1). The first human trials of the drug in healthy volunteers show a reduction in muscle glycogen (2). It is likely that the drug will eventually be useful in treating horses with Polysaccharide Storage Myopathy type 1 (PSSM1), resulting from a variant of glycogen synthase I (GYS1-R309H) that causes constitutive activation of GYS1. The work on Pompe disease is summarized in an editorial (3).
WHAT IS POMPE DISEASE?
Human Pompe Disease (GSD2) is a glycogen storage disease caused by loss-of-function alleles of GAA, the gene encoding the lysosomal alpha-1,4-glucosidase (4, 5). Patients with Pompe disease are able to synthesize glycogen, but are unable to break down glycogen as a source of energy in muscle tissue. Some alleles with 1% of enzyme activity or less are associated with juvenile-onset Pompe disease, characterized by cardiomyopathy and hypotonic skeletal muscle. Other alleles with higher levels of residual enzyme activity present as adult-onset disease primarily affecting skeletal muscle. Human Pompe Disease is a recessive disorder, meaning that both alleles of GAA must be defective in order for patients to show symptoms. OMIM gives an overview of clinical symptoms associated with Pompe Disease.
GLYCOGEN METABOLISM
Glycogen is a polymer of glucose that allows glucose to be stored for later use. In the liver, stored glycogen is converted to glucose and enters the bloodstream to maintain glucose homeostasis. In muscle and other tissues, stored glycogen is converted to glucose that is used as a source of energy. In some cases, the enzyme that catalyzes a particular reaction is encoded by different genes in liver and muscle. For example, GYS1, the gene affected by the GYS1-R309H variant associated with PSSM1 in horses, encodes the muscle form of glycogen synthase. The liver form of glycogen synthase is encoded by GYS2.
In this blog post, we will focus only on the enzymes required for glycogen synthesis (glycogenesis) and glycogen breakdown (glycogenolysis). Other enzymes are involved in the modification of glucose and are not discussed here.
Table 1 shows the genes encoding selected enzymes in human and horse. The human population is very large and well-studied; there are disease states associated with mutations in each of the genes shown in the table. In horse, which has a smaller population size and a much smaller research effort, disease states associated with mutations in two of the enzymes are known.
Table 1. Key enzymes responsible for glycogen synthesis and breakdown. The gene symbols are linked to the entries for the human genes in UniProt. Human diseases are named Glycogen Storage Disease (GSD) followed by a number. The human diseases are linked to the description in OMIM, while the horse diseases are linked to the description in OMIA.
Gene Enzyme name Human Disease Horse Disease
GYS1 Glycogen synthase, muscle GSD0, muscle PSSM1
GYS2 Glycogen synthase, liver GSD0, liver none
GBE1 Glycogen branching enzyme GSD4 GBED
AGL Glycogen debranching enzyme GSD3 none
GAA Glucosidase, alpha, acid GSD2 (Pompe disease) none
PYGM Glycogen phosphorylase, muscle GSD5 (McCardle disease) none
Figure 1 shows the process of glycogen synthesis and breakdown.
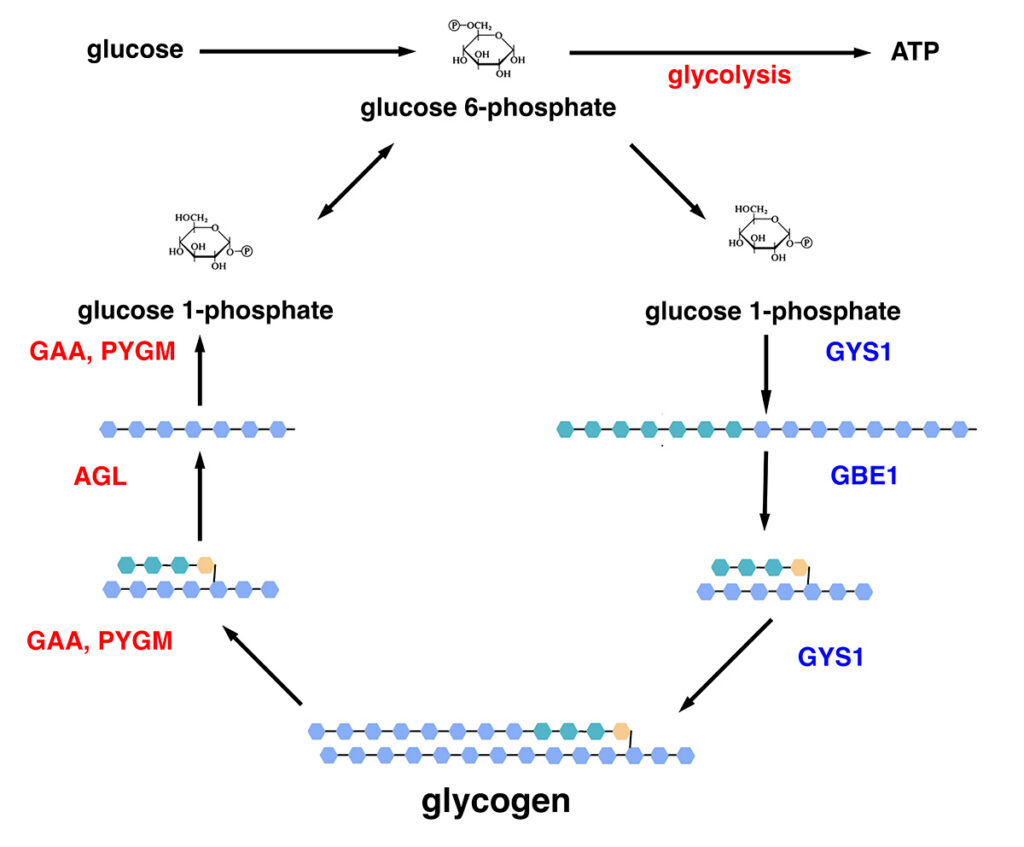
Figure 1. Synthesis (blue pathway) and breakdown (red pathway) of glycogen in muscle. Glucose is converted to glucose 6-phosphate. Under condition of abundant glucose and high energy demand, glucose is metabolized for energy through the multistep process of glycolysis, producing ATP. Under conditions of excess glucose and low energy demand, glucose is converted to glucose 1-phosphate, then to UDP-glucose (not shown). Glycogen synthase (GYS1) adds glucose units to growing glucose polymers using alpha-1,4 linkages. Branching enzyme (GBE1) creates branches by cleaving off segments of the growing polymer and adding them to the polymer using alpha-1,6 linkages. Mature glycogen contains hundreds of glucose units in a branched structure. Under conditions of low glucose and high energy demand, alpha-1,4-glucosidase (GAA) and glycogen phosphorylase (PYGM) cleave off glucose units as glucose 1-phosphate. Debranching enzyme (AGL) cleaves the alpha-1,6 linkages to allow cleavage of alpha-1,4 linkages to continue. Glucose 1-phosphate is converted to glucose 6-phosphate and enters glycolysis to produce energy.
Equine Polysaccharide Storage Myopathy, type 1 (PSSM1) is associated with a gain-of-function mutation in glycogen synthase, GYS1 (6). Glycogen synthase is the rate-limiting step in glycogenesis and is regulated post-translationally by phosphorylation. The GYS1-R309H allele associated with PSSM1 is constitutively activated and no longer inhibited by phosphorylation (7).
TREATMENT OF POMPE DISEASE
Enzyme replacement therapy: The current treatment for Pompe disease is infusion with alpha-1,4-glucosidase (GAA), the enzyme affected in Pompe disease. This treatment is only partially effective and loses efficacy over several years (8).
Gene therapy: In the long term, gene therapy may be the best treatment for human patients with Pompe disease (9). This work is in its early stages and is not relevant to horse.
Substrate reduction therapy: An emerging treatment for Pompe disease is substrate reduction therapy (1, 2). Pompe disease patients have a reduced ability to break down muscle glycogen, so a possible therapy is to inhibit GYS1, reducing the amount of muscle glycogen. This emerging treatment has relevance to horses with PSSM1, which have a constitutively active glycogen synthase (GYS1-R309H).
A DRUG FOR POMPE DISEASE?
Drug discovery: Substrate reduction therapy for Pompe disease requires a small molecule that inhibits GYS1 (expressed in muscle) but not GYS2 (expressed in liver). A high-throughput screen identified a small molecule, MZE001, that inhibits human GYS1 without inhibiting GYS2 (1). The drug appears to be a noncompetitive allosteric inhibitor of GYS1, with a binding site distinct from those for uridine diphosphate glucose (UDP glucose) and glucose-6-phosphate. The in vitro findings were confirmed by culturing primary fibroblasts from Pompe disease patients and normal controls. Fibroblasts from Pompe disease patients had elevated glycogen levels compared to controls; the levels were reduced when the cells were treated with MZE001 (1, 2).
Consequences of reduced GYS1 activity in patients: Before undertaking preclinical studies of MZE001 in laboratory animals, it is important to find out what happens to human patients with reduced GYS1 activity. The PPP1R3A gene encodes a protein that assists in transporting GYS1 to the correct location in the cell. Human patients with mutations that inactivate PPP1R3A have reduced activity of GYS1 and a reduction in muscle glycogen (10). Human patients with one copy of the variant have a 65% reduction in GYS1 activity (1). There is no obvious phenotype associated with having this level of reduction of muscle glycogen.
Mouse model of Pompe disease: A mouse model of Pompe disease was created through targeted mutation of the GAA gene (11, 12). Mice with an engineered knockout of GAA have a phenotype that replicates aspects of the human disease: excess glycogen in muscle tissue, enlarged lysosomes full of glycogen, and progressive loss of muscle strength.
The mouse model of Pompe disease has been used to investigate whether substrate limitation therapy targeting GYS1 might be useful for human patients. Both antisense oligonucleotides directed against GYS1 and an engineered knockout of GYS1 are effective at reducing glycogen accumulation in the mouse model of Pompe disease (13, 14).
The GYS1 inhibitor MZE001 was tested using the mouse model of Pompe disease (1, 2). Results show that MZE001 inhibition of GYS1 in Pompe disease mice reduces the amount of glycogen in muscle. The reduction in glycogen when Pompe disease mice are treated with MZE001 is also seen in primary fibroblasts and peripheral blood mononuclear cells (PBMCs). These are important findings for human studies, because a blood draw for PBMCs is less invasive than sampling skin for primary fibroblast culture, which is in turn less invasive than muscle biopsy.
Density-labeled glucose (13C6-glucose) was used to study the effects of MZE001 on in vivo glycogen synthesis without contamination by previously synthesized glycogen (1). Pompe disease mice incorporated about twice the amount of density-labeled glycogen compared to wild-type controls. MZE001 inhibited the synthesis of density-labeled glycogen in muscle in both Pompe mice and controls, without inhibiting glycogen synthesis in liver.
Administration of a pulse of density-labeled glucose allows the measurement of the half-life of newly-synthesized glycogen (1). In normal mice, the half-life of glycogen is about 16 hours, compared to several weeks in Pompe disease mice, suggesting that treatment with MZE001 would take several weeks to show maximal effect. Chronic treatment of normal mice with MZE001 showed reductions of glycogen at 4 and 14 weeks of 71% and 81%, respectively, while Pompe mice showed reductions of 38% and 58%. Efficacy of treatment was increased when used in conjunction with enzyme replacement therapy.
Drug trials on healthy human subjects: The GYS1 inhibitor MZE001was administered to healthy human subjects in a phase I clinical trial (2). The drug was found to be well tolerated in a dosage developed using the mouse model of Pompe disease. Subjects in the trial were evaluated using standard clinical tests, vital signs, EKGs, and exercise tolerance as evaluated using a treadmill test, with no abnormalities seen.
To evaluate the reduction in glycogen synthesis, most subjects were evaluated using PBMCs, established as useful using the mouse model of Pompe disease. A subset of subjects were evaluated using needle biopsies of muscle (2). Subjects showed a dose-dependent reduction in glycogen synthesis in PBMCs and muscle when treated with MZE001.
WHAT ABOUT HORSES?
Pompe disease is not the same as PSSM1: Pompe disease, resulting from a defect in GAA, the primary enzyme responsible for the breakdown of muscle glycogen, is distinct from equine Polysaccharide storage myopathy type 1 (PSSM1), resulting from a defect that constitutively activates GYS1, the rate-limiting enzyme for muscle glycogen synthesis. The pathology of Pompe disease includes a derangement of lysosome maturation as they become filled with glycogen that cannot be broken down. There is also an increase in the activity of GYS1 in Pompe disease, due to increases in glucose-6-phosphate (G6P), an allosteric activator of GYS1. The cytosolic enzyme glycogen phosphorylase (PYGM) appears to be incapable of clearing muscle glycogen in Pompe disease due to widespread disruptions in metabolism that affect this pathway as well.
Treatment of PSSM1 with MZE001: Will this drug become a treatment for horses with PSSM1? Horses with PSSM1 have one or two copies of a genetic variant in the gene encoding glycogen synthase I (GYS1-R309H). The variant causes the constitutive activation of glycogen synthase. The variant form of the enzyme is active even when phosphorylated, which normally reduces the activity of glycogen synthase (7).
The first experiments to evaluate MZE001 as a treatment for PSSM1 in horses are clear.
First, we need to measure the ability of MZE001 to inhibit both the normal and variant forms of equine glycogen synthase I. Maile et al. (7) produced both normal and variant forms of equine glycogen synthase for enzyme assays. MZE001 should be tested as an inhibitor of both forms as described (1).
MZE001 is a noncompetitive inhibitor of GYS1 in humans, mice, and dogs, suggesting that it would also inhibit equine GYS1 (1, 2). It is unknown whether MZE001 would inhibit constitutively activated GYS1-R309H. If MZE001 inhibits GYS1-R309H, it is a potential treatment for PSSM1.
Second, it would be helpful to measure the effects of MZE001 on glycogen levels in primary fibroblasts from horses of all three genotypes: n/n, n/P1, and P1/P1 horses. Taking a skin biopsy for the fibroblast assay is minimally invasive, and does not expose horses to the drug.
If these experiments show that MZE001 is effective at inhibiting equine GYS1-R309H, the next steps are clear. First, a small number of healthy horses should be treated with the drug to see if it is tolerated, and if glycogen is reduced in PBMCs or primary fibroblasts in horses treated with the drug. Finally, horses of all three genotypes should be studied to measure glycogen levels in PBMCs or primary fibroblasts, and finally, by muscle biopsy.
I only have access to public information on this subject and am not aware of any current unpublished research results on inhibition of equine GYS1-R309H by MZE001. I hope that this drug will ultimately prove effective at treating horses with PSSM1.
REFERENCES
1. Ullman JC, et al. (2024a) Small-molecule inhibition of glycogen synthase 1 for the treatment of Pompe disease and other glycogen storage disorders Sci Transl Med. 16(730):eadf1691. PMID: 38232139.
2. Ullman JC, et al. (2024b) First-in-human evaluation of safety, pharmacokinetics and muscle glycogen lowering of a novel glycogen synthase 1 inhibitor for the treatment of Pompe disease. Clin Pharmacol Ther. 116(6):1580-1592. PMID:39439155.
3. Koch RL, et al. (2024) Expanding therapeutic options for Pompe disease: a new small molecule inhibitor of glycogen synthase 1 (GYS1) shows preclinical promise in Pompe disease. Ann Transl Med. 12(6):123. PMID: 39817232.
4. Martiniuk, F et al. (1990) Identification of the base-pair substitution responsible for a human acid alpha glucosidase allele with lower ‘affinity’ for glycogen (GAA 2) and transient gene expression in deficient cells. Am. J. Hum. Genet. 47: 440-445. PMID: 2203258.
5. Wokke, JH et al. (1995) Genotype-phenotype correlation in adult-onset acid maltase deficiency. Ann. Neurol. 38: 450-454. PMID: 7668832.
6. McCue M, et al. (2008) Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 91(5):458-66. PMID: 18358695.
7. Maile CA, et al. (2017) A highly prevalent equine glycogen storage disease is explained by constitutive activation of a mutant glycogen synthase. Biochim Biophys Acta. 1861(1 Pt A): 3388–3398. PMID: 27592162.
8. Corsini A (2025) Improving the treatment of Pompe disease with enzyme replacement therapy: current strategies and clinical evidence. Expert Opin Pharmacother. 26(7):835-848. PMID: 40237692.
9. Leon-Astudillo C, et al. (2023) Current avenues of gene therapy in Pompe disease. Curr Opin Neurol. 36(5):464-473. PMID: 37639402.
10. Savage DB, et al. (2008) A prevalent variant in PPP1R3A impairs glycogen synthesis and reduces muscle glycogen content in humans and mice. PLoS Med. 5(1):e27. PMID: 18232732.
11. Raben N, et al. (1998) Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem. 273(30):19086-92. PMID: 9668092.
12. Taylor KM, et al. (2013) Dysregulation of multiple facets of glycogen metabolism in a murine model of Pompe disease. PLoS One. 28(2):e56181. PMID: 23457523.
13. Douillard-Guilloux G, et al. (2008) Modulation of glycogen synthesis by RNA interference: towards a new therapeutic approach for glycogenosis type II. Hum Mol Genet. 17(24):3876-86. PMID: 18782850.
14. Douillard-Guilloux G et al. (2010) Restoration of muscle functionality by genetic suppression of glycogen synthesis in a murine model of Pompe disease. Hum Mol Genet. 19(4):684-96. PMID: 19959526.
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
Carnegie Mellon University Profiles EquiSeq Founder
April 20, 2026 – Pittsburgh, Pennsylvania The alumni magazine for Carnegie Mellon published…
22 Apr 2026
EquiSeq hosts webinar on Recurrent Exertional Rhabdomyolysis (RER)
ALBUQUERQUE, NEW MEXICO – April 4, 2026 EquiSeq hosted a webinar on Recurrent…
4 Apr 2026
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
