
Blog
Peer Review Fails to Identify Errors in Inconclusive Study of Genetic Variants of Equine Muscle Genes
1 Jul 2020

This blog post is a discussion of a paper published by Williams et al. in a recent issue of Equine Veterinary Journal [1]. The paper aims to identify genetic variants that might be responsible for Myofibrillar Myopathy (MFM) in Warmbloods. The authors use a candidate gene approach, searching for variants of eight of the nine genes known to be associated with MFM in human patients and eight additional genes. Variants are identified by RNA-Seq of 16 warmbloods typed by muscle biopsy (8 affected and 8 control), and by analysis of additional whole genome sequence data available to the authors and in public databases.
The citation is:
Zoe J. Williams (will3084@msu.edu), Deborah Velez-Irizarry (no contact information), Jessica L. Petersen (jessica.petersen@unl.edu), Julien Ochala (julien.ochala@kcl.ac.uk), Carrie J. Finno (cjfinno@ucdavis.edu) and Stephanie J. Valberg (valbergs@cvm.msu.edu) (2020) Candidate gene expression and coding sequence variants in Warmblood horses with myofibrillar myopathy. Equine Vet J. 00: 1-10. PMID: 32453872.
Contents
- Summary
- Background
- Is Warmblood MFM inherited?
- PLEC and SQSTM1 are poor choices of candidate genes
- Identification of genetic variants
- Table 1
- Variants of FLNC and MYOT that are part of EquiSeq’s Myopathy Panel
- Test for association of genetic variants with Warmblood MFM
- Table 2
- Incidence of genetic variants among breeds
- Evaluation of genetic variants of FLNC and MYOT
- The P2 genetic variant of MYOT corresponds to S233P, not V238A
- REQUEST FOR PRINTED CORRECTION #1
- The amino acids affected by the equine V238A and S233P variants of MYOT do not differ among human MYOT isoforms
- Table 3
- Table 4
- REQUEST FOR PRINTED CORRECTION #2
- Conclusions
- References
Summary
Ninety variants were identified in exons of thirteen of the sixteen genes (ACTA1, BAG3, DES, DNAJB6, FLNC, HSPB8, KY, LDB3, LMNA, MYOT, PLEC, PYROXD1, and SQSTM1). No coding variants were found for CRYAB, FHL1, or TIA1. Of the 90 variants, 62 were synonymous substitutions, 2 were splice region variants, and 26 were missense alleles. All 26 missense alleles (in 11 genes) were predicted to have moderate effects using Ensembl’s Variant Effect Predictor (v98) [2].
The authors compared eight Warmbloods diagnosed with MFM by muscle biopsy with positive desmin stain to eight Warmbloods diagnosed as normal by muscle biopsy with negative desmin stain. None of the 26 missense alleles show a statistically significant difference in frequency between the Warmbloods diagnosed with MFM and the control Warmbloods. The 26 missense alleles include two variants of MYOT and FLNC that are offered as commercial tests by EquiSeq and the Center for Animal Genetics.
A trial of this size using unrelated human patients diagnosed with MFM would be unlikely to show a statistically significant association between the disease state and genetic variants of any of the eight human genes known to be associated with MFM. The results of this statistically underpowered study must be considered inconclusive.
In addition, the paper contains errors that were not identified during the process of peer review.
Background
Myofibrillar Myopathy (MFM) in humans is a neuromuscular disease state characterized by progressive muscular weakness. MFM is associated with specific morphological changes in skeletal muscle, resulting from disintegration of the sarcomeric Z disc and myofibrils. Affected muscle tissues shows ectopic aggregates of proteins that are structural components of the Z disc, including desmin (DES), alpha-B-crystallin (CRYAB), myotilin (MYOT), and filamin C (FLNC), as well as proteins involved in scavenging misfolded proteins targeted for proteolyis (BAG3). Mutations in nine genes (DES, CRYAB, MYOT, LDB3, FLNC, BAG3, KY, PYROXD1, and TTN) account for about half of the human cases of MFM, while the other half of cases cannot be assigned to one of these nine genes, suggesting that mutations in additional genes account for these cases (see [1] for references).
Other mutations in the nine genes known to be associated with MFM in human patients cause disease states that are different from MFM. These include cardiomyopathies with no apparent skeletal muscle involvement and myopathies lacking the characteristic morphological features of MFM.
The genes responsible for human cases of MFM were identified by analysis of kindreds displaying dominantly inherited Myofibrillar Myopathy. The first molecular cause, mutations in the gene encoding desmin (DES), was discovered in 1998 [3], with other genes being added to the list of causes as molecular techniques improved. Mutations in DES, MYOT, and LBD3 genes account for about half of the human cases where a molecular diagnosis can be made.
Is Warmblood MFM inherited?
Myofibrillar Myopathy (MFM) has been described in horses, both in Arabians [4] and Warmbloods [5]. In Warmbloods, horses from a three-generation kindred showing symptoms of MFM (exercise intolerance, a reluctance to move forward under saddle, and shifting lameness with no radiographic findings) show desmin-positive aggregates on muscle biopsy [5]. This work suggests autosomal dominant inheritance like that seen in most cases of human MFM.
It is therefore reasonable to suspect that missense alleles of genes associated with human MFM (DES, CRYAB, MYOT, LDB3, FLNC, BAG3, KY, PYROXD1, and TTN) might be associated with MFM in horses. For some reason, the authors do not include TTN in the list of candidate genes, despite the evidence that mutations in TTN are responsible for cases of MFM in several affected kindreds [6].
The other candidate genes chosen by the authors (FHL1, DNAJB6, PLEC, LMNA, ACTA1, HSPB8, SQSTM1, and TIA1) were chosen on the basis of characteristic findings on muscle biopsy of human patients that are somewhat similar to those seen in MFM (see [1] for references).
PLEC and SQSTM1 are poor choices of candidate genes
While there are certainly other candidate genes that might be added to the list, the inclusion of PLEC stands out as a particularly poor choice. A specific mutation in PLEC is associated with a recessive form of Limb-Girdle Muscular Dystrophy (LGMDR17) [7]. This mutation affects a specific exon of the PLEC gene, which serves as the first exon of a muscle-specific isoform of the PLEC protein [7]. Mutations elsewhere in the PLEC gene affect common exons shared with isoforms expressed in skin. Mutations affecting isoforms expressed in skin are associated with forms of epidermolysis bullosa [8, 9, 10, 11, 12], a blistering disease that would be conspicuous were it to occur in horses.
Similarly, while one allele of SQSTM1 is associated with a myopathy in human patients [13], the majority of mutant alleles of SQSTM1 identified in human patients are associated with Paget disease of bone, frontotemporal dementia, or neurodegeneration, with no evidence of myopathy [14, 15, 16, 17, 18, 19, 20, 21].
Identification of genetic variants
Table 1 (adapted from supplementary data in [1]) shows the 90 genetic variants considered in further detail. The table shows variant positions in EquCab 3.0 and EquCab 2.0 (some variants cannot be positioned in the less-complete assembly EquCab 2.0), the SNP (reference/variant, note that these are sometimes reversed between EquCab 3.0 and EquCab 2.0), the codon number and amino acids for reference and variant, and the type of variant (synonymous, missense, or splice).
| Table 1. Variants Reported in Horsesa | |||||||
|---|---|---|---|---|---|---|---|
| Gene | EquCab3b | SNP 3.0c | EquCab2b | Chromosomed | SNP 2.0c | Variant EquCab 3.0e | Typef |
| BAG3 | chr1:12965712 | C/T | chr1:12853224 | chr1 | C/T | R522R | Synonymous |
| BAG3 | chr1:12966009 | C/T | chr1:12853521 | chr1 | C/T | P423P | Synonymous |
| BAG3 | chr1:12966053 | C/T | chr1:12853565 | chr1 | C/T | A409T | Missense |
| BAG3 | chr1:12966156 | C/T | chr1:12853668 | chr1 | C/T | P374P | Synonymous |
| BAG3 | chr1:12971976 | A/G | chr1:12859493 | chr1 | A/G | S114S | Synonymous |
| ACTA1 | chr1:68960019 | G/C | chr1:68409837 | not present | G17G | Synonymous | |
| ACTA1 | chr1:68961415 | C/T | chr1:68411233 | not present | G304G | Synonymous | |
| LDB3 | chr1:84589937 | T/C | chr1:83735063 | chr1 | T/C | I590V, I583V, I496V | Missense |
| LDB3 | chr1:84590004 | G/A | chr1:83735130 | chr1 | G/A | D567D, D560D, D473D | Synonymous |
| LDB3 | chr1:84597964 | A/G | chr1:83743186 | chr1 | A/G | A360A, A353A, A266A | Synonymous |
| LDB3 | chr1:84618893 | C/T | chr1:83764115 | chr1 | C/T | E219E, E120E, E219E | Synonymous |
| LDB3 | chr1:84626133 | A/G | chr1:83771402 | chr1 | A/G | L48L | Synonymous |
| FLNC | chr4:83825648 | C/T | chr4:83724118 | not present | D236D | Synonymous | |
| FLNC | chr4:83839914 | G/C | chr4:83738384 | chr4 | G/C | A1316A, A1295A | Synonymous |
| FLNC | chr4:83840299 | G/A | chr4:83738769 | chr4 | G/A | A1445T, A1424T | Missense |
| FLNC | chr4:83845892 | C/T | chr4:83744362 | chr4 | C/T | Splice? | |
| FLNC | chr4:83846148 | G/A | chr4:83744618 | chr4 | G/A | P2064P, P2091P, P2070P, P2062P | Synonymous |
| FLNC | chr4:83847237 | C/T | chr4:83745707 | chr4 | C/T | S2181S, S2208S, S2187S, S2179S | Synonymous |
| DNAJB6 | chr4:107802363 | G/T | chr4:107686038 | chr4 | G/T | A249S, A134S | Missense |
| LMNA | chr5:38652136 | A/C | chr5:42041844 | chr5 | A/C | D447E, D559E, D475E | Missense |
| LMNA | chr5:38654588 | T/G | chr5:42044296 | chr5 | T/G | R168R, R280R, R196R | Synonymous |
| LMNA | chr5:38667865 | C/T | chr5:42057573 | chr5 | C/T | V69V | Synonymous |
| DES | chr6:8696183 | T/G | chr6:8923475 | not present | S31A | Missense | |
| DES | chr6:8696317 | A/C | chr6:8923609 | chr6 | A/C | P75P, P33P | Synonymous |
| DES | chr6:8701939 | G/A | chr6:8929231 | chr6 | G/A | G485G | Synonymous |
| PYROXD1 | chr6:48903253 | G/A | chr6:47640369 | chr6 | A/G | A30A | Synonymous |
| PYROXD1 | chr6:48903284 | A/G | chr6:47640400 | chr6 | A/G | I41V | Missense |
| PYROXD1 | chr6:48917278 | A/G | chr6:47654417 | chr6 | G/A | A204A, A189A, A146A | Synonymous |
| PYROXD1 | chr6:48918053 | G/A | chr6:47655192 | chr6 | A/G | R233K, R218K, R175K | Missense |
| PYROXD1 | chr6:48918068 | G/A | chr6:47655207 | chr6 | A/G | R238Q, R223Q, R180Q | Missense |
| PYROXD1 | chr6:48920320 | C/T | chr6:47657549 | chr6 | C/T | C327C, C312C, C269C | Synonymous |
| PYROXD1 | chr6:48924733 | T/C | chr6:47661961 | chr6 | C/T | Y501Y, Y486Y, Y445Y, Y443Y | Synonymous |
| PYROXD1 | chr6:48924749 | G/C | chr6:47661977 | chr6 | G/C | D507H, D492H, D451H, D449H | Missense |
| HSPB8 | chr8:17051982 | G/A | chr8:14656786 | chr8 | G/A | H39H | Synonymous |
| PLEC | chr9:84699001 | G/T | chr9:82497996 | chr9 | G/T | S4510S, S4474S, S4581S, S4625S, S4451S, S4483S, S4487S | Synonymous |
| PLEC | chr9:84699013 | G/A | chr9:82498008 | chr9 | G/A | S4506S, S4470S, S4577S, S4621S, S4447S, S4479S, S4483S | Synonymous |
| PLEC | chr9:84699187 | G/A | chr9:82498183 | chr9 | G/A | T4448T, T4412T, T4519T, T4563T, T4389T, T4421T, T4425T | Synonymous |
| PLEC | chr9:84699202 | C/T | chr9:82498198 | chr9 | C/T | T4443T, T4407T, T4514T, T4558T, T4384T, T4416T, T4420T | Synonymous |
| PLEC | chr9:84699481 | G/A | chr9:82498477 | chr9 | G/A | D4350D, D4314D, D4421D, D4465D, D4391D, D4323D, D4327D | Synonymous |
| PLEC | chr9:84699496 | G/A | chr9:82498492 | chr9 | G/A | T4345T, T4309T, T4416T, T4460T, T4266T, T4318T, T4322T | Synonymous |
| PLEC | chr9:84700174 | G/A | chr9:82499170 | chr9 | G/A | D4119D, D4083D, D4190D, D4234D, D4060D, D4092D, D4096D | Synonymous |
| PLEC | chr9:84701209 | G/A | chr9:82500205 | not present | A3774A, A3738A, A3845A, A3889A, A3715A, A3747A, A3751A | Synonymous | |
| PLEC | chr9:84701301 | C/T | chr9:82500297 | chr9 | C/T | A3744T, A3708T, A3815T, A3859T, A3685T, A3717T, A3721T | Missense |
| PLEC | chr9:84701459 | C/T | chr9:82500455 | chr9 | C/T | S3691N, S3655N, S3762N, S3806N, S3632N, S3664N, S3668N | Missense |
| PLEC | chr9:84701931 | C/T | chr9:82500927 | chr9 | C/T | D3534N, D3498N, D3605N, D3649N, D3475N, D3507N, D3511N | Missense |
| PLEC | chr9:84702701 | G/A | chr9:82501697 | chr9 | G/A | T3277M, T3241M, T3348M, T3392M, T3218M, T3250M, T3254M | Missense |
| PLEC | chr9:84702893 | A/G | chr9:82501889 | chr9 | A/G | V3213A, V3177A, V3284A, V3328A, V3154A, V3186A, V3190A | Missense |
| PLEC | chr9:84703198 | G/A | chr9:82502194 | chr9 | G/A | S3111S, S3075S, S3182S, S3226S, S3052S, S3084S, S3088S | Synonymous |
| PLEC | chr9:84703213 | G/A | chr9:82502209 | chr9 | G/A | D3106D, D3070D, D3177D, D3221D, D3047D, D3079D, D3083D | Synonymous |
| PLEC | chr9:84703327 | G/A | chr9:82502323 | not present | Y3068Y, Y3032Y, Y3139Y, Y3183Y, Y3009Y, Y3041Y, Y3045Y | Synonymous | |
| PLEC | chr9:84703372 | G/A | chr9:82502368 | chr9 | G/A | I3053I, I3017I, I3124I, I3168I, I2994I, I3026I, I3030I | Synonymous |
| PLEC | chr9:84703445 | C/T | chr9:82502441 | chr9 | C/T | R3029Q, R2993Q, R3100Q, R3144Q, R2970Q, R3002Q, R3006Q | Missense |
| PLEC | chr9:84703610 | G/A | chr9:82502606 | chr9 | G/A | T2974M, T2938M, T3045M, T3089M, T2915M, T2947M, T2951M | Missense |
| PLEC | chr9:84704059 | G/A | chr9:82503055 | chr9 | G/A | I2824I, I2788I, I2895I, I2765I, I2939I, I2797I, I2801I | Synonymous |
| PLEC | chr9:84704134 | A/G | chr9:82503130 | chr9 | A/G | R2799R, R2763R, R2870R, R2914R, R2740R, R2772R, R2776R | Synonymous |
| PLEC | chr9:84704450 | T/C | chr9:82503446 | chr9 | T/C | E2694G, E2658G, E2765G, E2809G, E2635G, E2667G, E2671G | Missense |
| PLEC | chr9:84704731 | C/A | chr9:82503727 | chr9 | C/A | L2600L, L2564L, L2671L, L2715L, L2541L, L2573L, L2577L | Synonymous |
| PLEC | chr9:84705316 | C/T | chr9:82504312 | chr9 | C/T | R2446H, R2410H, R2517H, R2561H, R2387H, R2419H, R2423H | Missense |
| PLEC | chr9:84705402 | A/G | chr9:82504398 | chr9 | A/G | A2417A, A2381A, A2488A, A2532A, A2358A, A2390A, A2394A | Synonymous |
| PLEC | chr9:84705954 | A/G | chr9:82504950 | chr9 | A/G | D2233D, D2197D, D2304D, D2348D, D2174D, D2206D, D2210D | Synonymous |
| PLEC | chr9:84706026 | C/T | chr9:82505022 | chr9 | C/T | A2209A, A2173A, A2280A, A2324A, A2150A, A2182A, A2186A | Synonymous |
| PLEC | chr9:84707250 | G/A | chr9:82506430 | chr9 | G/A | A1801A, A1765A, A1872A, A1916A, A1742A, A1774A, A1778A | Synonymous |
| PLEC | chr9:84708309 | C/T | chr9:82507489 | chr9 | C/T | E1448E, E1412E, E1519E, E1563E, E1389E, E1421E, E1425E | Synonymous |
| PLEC | chr9:84708435 | G/A | chr9:82507615 | not present | H1406H, H1370H, H1477H, H1521H, H1347H, H1379H, H1383H | Synonymous | |
| PLEC | chr9:84710460 | C/T | chr9:82509640 | chr9 | C/T | Q1192Q, Q1156Q, Q1263Q, Q1307Q, Q1133Q, Q1165Q, Q1169Q | Synonymous |
| PLEC | chr9:84711951 | G/A | chr9:82511131 | chr9 | G/A | P1088P, P1052P, P1159P, P1203P, P1029P, P1061P, P1065P | Synonymous |
| PLEC | chr9:84714901 | A/G | chr9:82514274 | chr9 | A/G | C739C, C703C, C810C, C854C, C680C, C712C, C716C | Synonymous |
| PLEC | chr9:84715705 | G/A | chr9:82515078 | chr9 | G/A | G615G, G579G, G686G, G730G, G556G, G588G, G592G | Synonymous |
| PLEC | chr9:84715932 | C/A | chr9:82515305 | not present | G568C, G532C, G639C, G683C, G509C, G541C, G545C | Missense | |
| PLEC | chr9:84717317 | G/T | chr9:82516690 | chr9 | G/T | R412R, R376R, R483R, R527R, R353R, R385R, R389R | Synonymous |
| SQSTM1 | chr14:1898112 | C/T | chr14:2658554 | chr14 | C/T | P438P | Synonymous |
| SQSTM1 | chr14:1899276 | G/A | chr14:2659718 | chr14 | G/A | P374S | Missense |
| SQSTM1 | chr14:1899799 | C/T | chr14:2660241 | chr14 | C/T | P296P | Synonymous |
| MYOT | chr14:37814680 | G/C | chr14:38515040 | not present | T433S | Missense | |
| MYOT | chr14:37818779 | C/T | chr14:38519139 | chr14 | C/T | K427K | Synonymous |
| MYOT | chr14:37818807 | A/G | chr14:38519167 | chr14 | A/G | V238A | Missense |
| MYOT | chr14:37818823 | A/G | chr14:38519183 | chr14 | A/G | S233P | Missense |
| MYOT | chr14:37822219 | T/G | chr14:38522579 | chr14 | T/G | Splice? | |
| MYOT | chr14:37826677 | C/T | chr14:38527037 | chr14 | C/T | K8K | Synonymous |
| KY | chr16:71625764 | A/G | chr16:70042017 | chr16 | A/G | A591A, A570A, A531A | Synonymous |
| KY | chr16:71626145 | C/T | chr16:70042398 | chr16 | C/T | T464T, T443T, T404T | Synonymous |
| KY | chr16:71626403 | C/T | chr16:70042656 | chr16 | C/T | T378T, T357T, T318T | Synonymous |
| KY | chr16:71631197 | G/A | chr16:70047450 | chr16 | G/A | S262S, S241S, S202S | Synonymous |
| KY | chr16:71638960 | C/A | chr16:70055284 | chr16 | C/A | A203A, A182A, A143A | Synonymous |
| KY | chr16:71661955 | C/G | chr16:70078279 | chr16 | C/G | L65L | Synonymous |
| KY | chr16:71661994 | G/A | chr16:70078318 | chr16 | G/A | N52N | Synonymous |
| KY | chr16:71665337 | C/T | chr16:70081660 | chr16 | A/C | R23Q | Missense |
| SQSTM1 | chr14:1910156 | G/A | NULL | A52A | Synonymous | ||
| SQSTM1 | chr14:1910307 | G/A | NULL | A2V | Missense | ||
| BAG3 | chr1:12988510 | C/T | NULL | L24L | Synonymous | ||
aAdapted from supplementary Table S1.
bChromosome and genomic coordinate for variant base in EquCab3.0 and EquCab 2.0.
cReference allele and variant allele. Note that these may differ between EquCab3.0 and EquCab2.0.
dChromosome is EquCab2.0; if site is missing from the assembly, it is noted as “not present.”
eAmino acid for the reference allele, codon positions for various models, and amino acid for the variant allele.
fAllele type: synonymous substitution (Synonymous), missense allele (Missense), or variant in intron near splice junction (Splice).
There is no documented instance of a synonymous substitution in any of the sixteen candidate genes being associated with a disease state in humans, so the 62 synonymous substitutions described here will not be considered further. Neither of the two potential splice site variants affect the most conserved splice donor or acceptor positions and will not be considered further here. There are 26 missense alleles worth considering.
Variants of FLNC and MYOT that are part of EquiSeq’s Myopathy Panel
It is notable that this study failed to identify one of two filamin C (FLNC) variants present as a haplotype, nicknamed P3. The P3 haplotype of FLNC consists of two missense alleles: P3a (chr4:83,837,774 G/A in EquCab3.0) and P3b (chr4:83,840,299 G/A in EquCab3.0). The P3a allele is a substitution of lysine (K) for glutamine (E) in a highly-conserved position in the FLNC protein, while the P3b allele is a substitution of threonine (T) for alanine (A) in a highly-conserved position in the FLNC protein. Both alleles are extensively described in the patent cited in this study (patent #WO2017165733A1), although the authors make no mention of the reason for their failure to identify the P3a variant, which must have been present in the horses from which they derived their sequence data. The failure to identify the P3a variant of FLNC in this study highlights the possibility that other potentially damaging alleles of the candidate genes were missed.
The images below shows traces from Sanger DNA sequencing identifying the P3a and P3b variants, as presented in the patent cited by the authors (patent #WO2017165733A1).
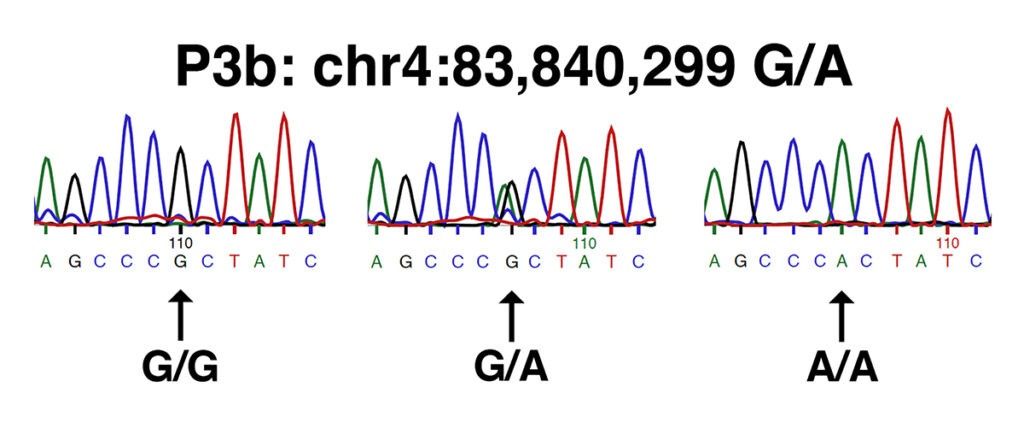
Both sets of traces show the sequence of the forward strand amplified from (left to right): wild type (n/n or G/G), a heterozygote (n/P3 or G/A), and a homozygote (P3/P3 or A/A).
This study identifies the P2 allele of MYOT (chr14:37,818,823 A/G in EquCab3.0), also described in the patent cited in this study (patent #WO2017165733A1). The P2 allele of MYOT is the substitution of a proline (P) for a serine (S) in a highly-conserved position in the serine-rich region of the MYOT protein. The image below shows traces from Sanger DNA sequencing identifying the P2 allele of MYOT, as presented in the patent cited by the authors (patent #WO2017165733A1).
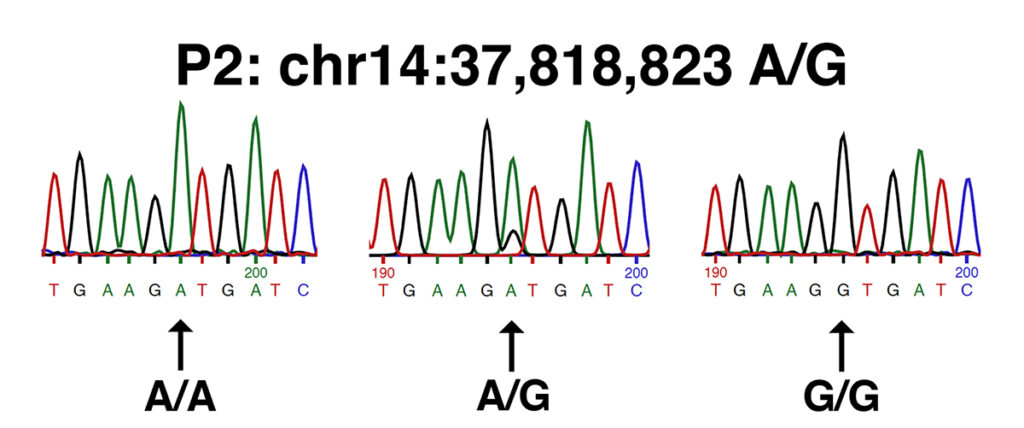
This set of traces shows the sequence of the forward strand amplified from (left to right): wild type (n/n or A/A), a heterozygote (n/P2 or A/G), and a homozygote (P2/P2 or G/G).
Test for association of genetic variants with Warmblood MFM
The authors attempt to find an association between genetic variants described in this study and Myofibrillar Myopathy (MFM) in Warmbloods. They select eight Warmbloods diagnosed with MFM by the presence of desmin aggregates in muscle tissue. These eight Warmbloods were selected for the availability of snap frozen samples of gluteus medius muscle. The authors make no statement about the relatedness of these eight MFM Warmbloods, so it is unlikely that any are directly related. A set of eight control Warmbloods was selected on the basis of a lack of symptoms, no evidence of desmin-positive aggregates in muscle tissue, and availability of snap frozen gluteus medius muscle.
The authors find no statistically significant association between any genetic variant described in this study and the MFM phenotype. Genotypes for the sixteen horses for 26 missense alleles identified in this study are shown in the table below (adapted from Figure 2 in [1]).
| Table 2. Variants in MFM and Control Warmbloodsa | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Variantb | Variant Proteinc | Warmbloods with MFMd | Control Warmbloodse | ||||||||||||||
| MFM WB1 | MFM WB2 | MFM WB3 | MFM WB4 | MFM WB5 | MFM WB6 | MFM WB7 | MFM WB8 | WT WB1 | WT WB2 | WT WB3 | WT WB4 | WT WB5 | WT WB6 | WT WB7 | WT WB8 | ||
| chr1:12966053 C/T | BAG3-A409T | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/n | n/n | n/n |
| chr1:84589937 T/C | LDB3-I590V | n/n | n/v | v/v | n/v | n/n | n/n | n/n | n/n | n/v | n/v | n/n | n/n | n/n | n/n | n/n | n/n |
| chr4:83840299 G/A | FLNC-A1424T (P3b) | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n |
| chr4:107802363 G/T | DNAJB-A249S | v/v | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/v | n/v | n/n | n/n | n/n | n/n | n/n | n/v |
| chr5:38652136 A/C | LMNA-D447E | v/v | n/v | n/v | n/v | n/v | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/v | v/v | n/n |
| chr6:8696183 T/G | DES-S31A | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/v | n/n | n/n | n/n | n/n |
| chr6:48903284 A/G | PYROXD1-I41V | v/v | v/v | n/v | v/v | v/v | n/v | v/v | v/v | v/v | v/v | n/v | n/v | v/v | v/v | v/v | v/v |
| chr6:48918053 G/A | PYROXD1-R233K | n/v | v/v | n/v | n/v | v/v | n/n | n/n | n/n | n/v | v/v | n/v | n/v | n/v | n/v | n/n | n/v |
| chr6:48918068 G/A | PYROXD1-R238Q | n/v | n/v | n/v | n/v | v/v | n/n | n/n | n/n | n/v | n/n | n/v | v/v | n/v | n/n | n/n | n/n |
| chr6:48924749 G/C | PYROXD1-D492H | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/v | n/n | n/n | n/n | n/n |
| chr9:84701301 C/T | PLEC-A3744T | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/v | n/v | n/n | n/n | n/n | n/n |
| chr9:84701459 C/T | PLEC-S3691N | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/n |
| chr9:84701931 C/T | PLEC-D3534N | n/n | n/n | n/n | n/n | n/v | n/n | n/v | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n |
| chr9:84702701 G/A | PLEC-T3277M | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n |
| chr9:84702893 A/G | PLEC-V3213A | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/n | n/n | n/n |
| chr9:84703445 C/T | PLEC-R3029Q | n/v | n/n | n/v | v/v | n/n | n/v | n/v | n/n | n/v | n/v | n/v | n/v | n/v | n/v | n/n | v/v |
| chr9:84703610 G/A | PLEC-T2974M | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v |
| chr9:84704450 T/C | PLEC-E2694G | n/n | n/n | n/n | n/v | n/v | n/v | v/v | n/v | n/n | n/v | n/n | n/v | n/v | n/n | n/n | n/v |
| chr9:84705316 C/T | PLEC-R2446H | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/v | n/n | n/n | n/n | n/n | n/n |
| chr9:84715932 C/A | PLEC-G568C | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/v | n/n | n/n | n/n |
| chr14:1899276 G/A | SQSTM1-P374S | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | v/v | n/n | n/n | n/n |
| chr14:1910307 G/A | SQSTM1-A2V | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | |
| chr14:37814680 G/C | MYOT-T433S | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n |
| chr14:37818807 A/G | MYOT-V238A | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/v | n/n | n/n | n/n | n/n | n/v | n/n | n/n |
| chr14:37818823 A/G | MYOT-S233P (P2) | n/n | n/v | n/n | v/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/v |
| chr16:71665337 C/T | KY-R23Q | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/n | n/v | n/n | n/n | n/n | n/n | n/n | n/n | n/n |
aAdapted from Figure 2 in [1].
bVariants identified by chromosomal location in EquCab3.0 and by reference/variant allele.
cAmino acid change produced by the missense allele. When several different protein models are given [see Table 1], one is selected to give the position of the amino acid substitution.
dWarmbloods scored with MFM by desmin staining of muscle biopsies.
eControl Warmbloods (wild-type or WT) scored by desmin staining of muscle biopsies.
It is a crude measure of the lack of statistical power of the analysis of this small sample that, for example, one of the PLEC variants (chr9:84,701,931 in EquCab3.0) is heterozygous in three of the MFM Warmbloods and entirely absent from the control Warmbloods, yet does not achieve statistical significance. As noted above, it is very unlikely that this variant is of any biological significance, as pathogenic alleles of PLEC not associated with the first exon of the muscle-specific isoform cause forms of epidermolysis bullosa, a skin-blistering phenotype that would scarcely escape notice.
The P2 allele of MYOT (chr14:37,818,823 in EquCab3.0) is homozygous in one of the MFM Warmbloods and heterozygous in another; it is heterozygous in two of the unaffected Warmbloods. This result is also of no statistical significance.
The P3b allele of FLNC (chr4:83,840,299 in EquCab3.0) is heterozygous in two of the MFM Warmbloods and absent from the unaffected Warmbloods. This result is also of no statistical significance.
Rather than discussing the frequency of individual variants in the eight MFM and eight control Warmbloods in this study, as above, the authors state:
…at most 5 of 8 MFM horses were heterozygous or homozygous for 1 of 5 variants, however, between 3 and 7 of the 8 non-MFM WB were heterozygous or homozygous for these same variants.
This generalization, however, is not true of one of the PLEC variants (chr9:84,701,931 in EquCab3.0), which as pointed out above, is heterozygous in three of the eight MFM horses and absent from all eight of the non-MFM horses.
It is worth pointing out that a test of this kind would almost certainly fail to identify genetic variants associated with MFM in humans in a study carried out in this way at this scale. All of the eight MFM genes identified in human patients whose equine orthologs are analyzed in this study were identified by first locating kindreds of affected patients, then analyzing affected and unaffected individuals from such kindreds. Eight unrelated human patients with a diagnosis of MFM by symptoms and histopathology of muscle samples would likely have diverse genetic defects. On average, half of human patients diagnosed with MFM do not receive a molecular diagnosis; of those that do, about half of the mutations are in three genes, DES, MYOT, and LBD3. Assuming that there were no pathogenic mutations in eight human controls, analysis of these sixteen samples would likely lack sufficient statistical power to identify any of the human MFM genes, although the allele frequency of pathogenic MFM alleles is low in human populations.
It would have been more productive to analyze related Warmbloods from, for example, the three-generation kindred showing apparent transmission of MFM [5], including both affected and unaffected Warmbloods in the study. Alternatively, study of a much larger sample of affected and control Warmbloods that were not directly related would also have been more productive. We await the results of studies of this kind in the future.
Incidence of genetic variants among breeds
The authors survey the incidence of the missense variants across breeds. Some of the missense variants display significant differences in the allele frequencies among breeds, while others do not. It is unclear what bearing these results might have on the identification of genetic variants associated with MFM in Warmbloods.
Evaluation of genetic variants of FLNC and MYOT
As noted above, the authors failed to identify one of the two FLNC variants that is part of the P3 haplotype. While identifying the P3b allele of FLNC (chr4:838,40,299 G/A in EquCab3.0), they failed to identify the P3a allele of FLNC (chr4:83,837,774 G/A in EquCab3.0), although it was certainly present in their samples.
Of more serious concern is an error in the discussion section of their paper. While correctly identifying the P2 alle of MYOT (chr14:37,818,823 A/G in EquCab3.0), they incorrectly identify the amino acid substitution caused by this allele. As shown above, while the P2 allele of MYOT (chr14:37,818,823 A/G in EquCab3.0) causes a substitution of a proline (P) for a serine (S) in codon 233 of the serine-rich region of the MYOT protein (S233P), in the discussion, the authors incorrectly assign the amino acid substitution caused by a different allele of MYOT (chr14:37,818,807 A/G in EquCab3.0) to the P2 allele. The allele of MYOT that they discuss (chr14:37,818,807 A/G in EquCab3.0), causes the V238A substitution. This is a benign substitution, as they note.
We have written to the editor of the Equine Veterinary Journal to request that a written correction be published to address this error, which escaped peer review.
The P2 genetic variant of MYOT corresponds to S233P, not V238A
It is easy for anyone to verify that the P2 allele of MYOT (chr14:37,818,823 A/G in EquCab3.0) causes the S233P substitution rather than the V238A substitution.
1. Go to:
http://genome.ucsc.edu
2. In the top menu bar, select “Genomes” to go to:
http://genome.ucsc.edu/cgi-bin/hgGateway
3. In the text box that says “Enter species or common name,” enter “Horse.”
4. Under “Find position,” select as the horse assembly “Jan. 2018 (EquCab3.0/equCab3)”
5. Under “Position/Search term” enter:
chr14:37,818,823
6. Click “GO.” This takes you to the Genome Browser. At the top of the screen, to the right of “Zoom Out,” click 3X twice.
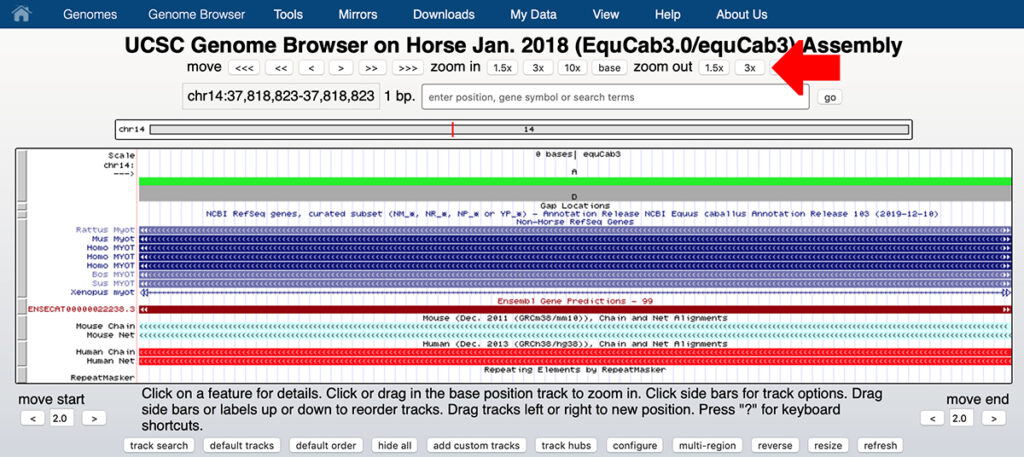
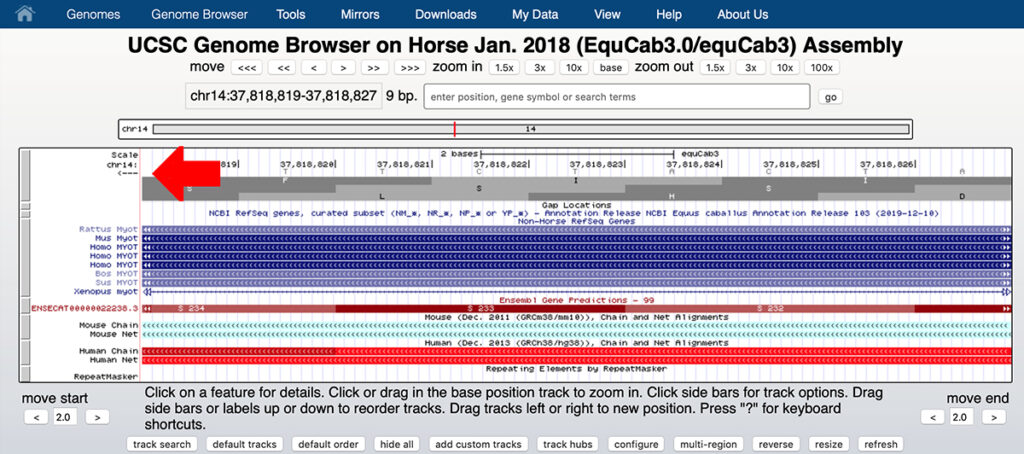
7. You should see a screen that resembles the screen shown below. Click the arrow at the top left to see the reverse strand rather than the forward strand as shown.
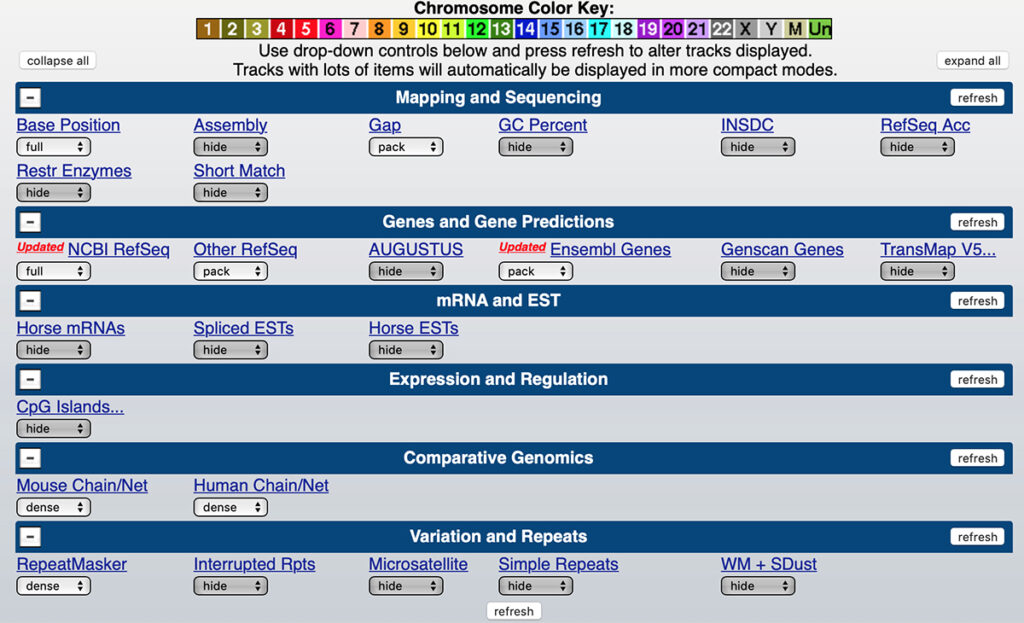
8. If your display is substantially different from that shown here, set the Genome Browser settings as shown below.
9. You can see that chr14:37,818,823 on the reverse strand is T, corresponding to the reference allele A on the forward strand.
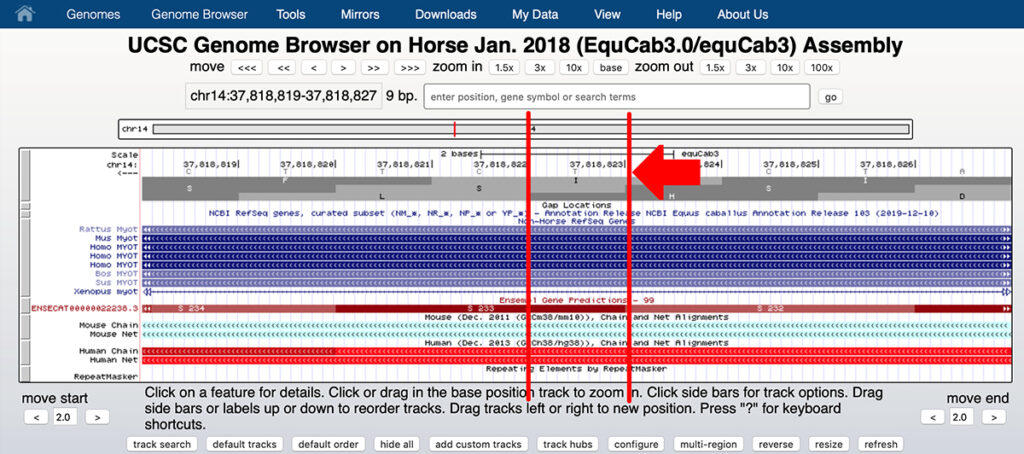
This is the first base of the serine codon S233. A substitution of G for A on the forward strand will change the base on the reverse stand from T to C, changing the codon from TCT (serine or S) to CCT (proline or P). You need to read the bases in the reverse strand from right to left.
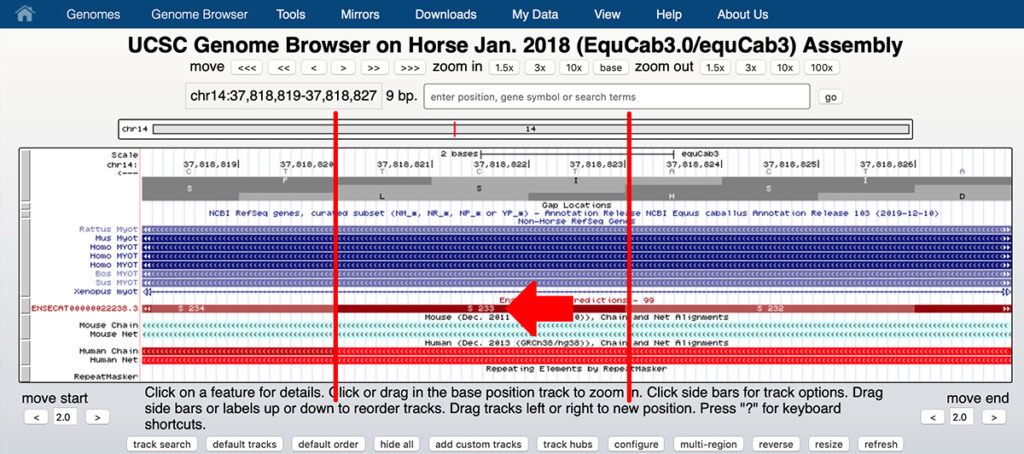
REQUEST FOR PRINTED CORRECTION: Therefore it is evident that chr14:37,818,823 A/G refers to a polymorphism in the first base of serine codon 233, changing it to proline 233, and that the allele is correctly described as MYOT-S233P, not V238A as described in the discussion section of the paper. We request that the Equine Veterinary Journal print a correction to this article [1].
The amino acids affected by the equine V238A and S233P variants of MYOT do not differ among human MYOT isoforms
The discussion [1
] contains the statement:
The V238A MYOT variant is predicted to be “tolerated” by SIFT classification and the same position in human MYOT encodes 1 of 3 different amino acids (N238, T238 and Y238) depending on isoform expression. Thus, this region is either not conserved across species or there are other isoforms not yet annotated in the current version of the equine genome.
This statement is not supported by any evidence on which database was used to assess human MYOT isoforms, the accession IDs of the protein sequences used to make this comparison, or the methods used in the analysis. One of the purposes of publication in a peer-reviewed journal is to permit other investigators to replicate the work described. As the authors do not cite any original work of their own on human MYOT isoforms or claim access to any private resources, it is reasonable to assume that they have based this statement on publicly available data from NCBI.
The canonical sequence for equine MYOT is a full-length MYOT protein having 498 amino acids (XP_014586147.1). This protein sequence aligns to the canonical human MYOT sequence (isoform a) having 498 amino acids (NP_006781.1). Two different human MYOT isoforms align to isoform a: isoform c (NP_001287840.1), with a length of 383 amino acids that is missing the N-terminal 115 amino acids, and isoform b (NP_001129412.1), with a length of 314 amino acids that is missing the N-terminal 184 amino acids.
There is also an isoform X1 with an alternative splice that alters the amino acid sequence N-terminal to amino acid 229 of isoform a, represented by three identical protein sequences (XP_016865549.1, XP_016865550.1, and XP_016865551.1). The evidence for these isoforms is computational; there does not appear to be experimental confirmation.
Table 3 compares the sequence of the equine MYOT protein to the sequence of the human MYOT protein around the site of the V238A variant. Eight human protein sequence IDs, representing four different protein sequences, are compared following sequence alignment. The sequence of the four different human sequences represented by eight entries at NCBI are identical at the variant site. The sequences of the ten amino acids N-terminal to the variant site are identical in isoforms a, b, and c, and are also identical to the equine sequence, but differ from the sequence in isoform X1. The sequences of the ten amino acids C-terminal to the variant site are identical among all isoforms and are identical to the equine sequence.
There is therefore no support for the author’s statement that the amino acid at the position of the V238A equine variant differs among human isoforms.
The substitution of an alanine (A) for a valine (V) is a conservative substitution, as noted by the authors [1], but as noted above, this is not the MYOT variant corresponding to chr14:37,818,823 A/G described in the patent cited by the authors (patent #WO2017165733A1), which is the basis of the commercial test.
| Table 3. Human isoforms of MYOT compared to horse MYOT near chr14:37,818,807 A/Ga | |||
|---|---|---|---|
| Accession IDb | Length (aa)c | Notesd | Sequence around equine V238A variante |
| XP_014586147.1 | 498 | Full length horse protein sequence | 227-VRSRSSSRGDVNDQDAIQEKF-247 |
| Q9UBF9.2 | 498 | isoform a: NP_006781.1, BAG35604.1, and Q9UBF9.2 are identical | 227-VRSRSTSRGDVNDQDAIQEKF-247 |
| BAG35604.1 | 498 | isoform a: NP_006781.1, BAG35604.1, and Q9UBF9.2 are identical | 227-VRSRSTSRGDVNDQDAIQEKF-247 |
| NP_006781.1 | 498 | isoform a: NP_006781.1, BAG35604.1, and Q9UBF9.2 are identical | 227-VRSRSTSRGDVNDQDAIQEKF-247 |
| NP_001287840.1 | 383 | isoform c: N-terminal 115 aa sequence missing vs. NP_006781.1 | 112-VRSRSTSRGDVNDQDAIQEKF-132 |
| NP_001129412.1 | 314 | isoform b: N-terminal 184 aa sequence missing vs. NP_006781.1 | 43-VRSRSTSRGDVNDQDAIQEKF-63 |
| XP_016865549.1 | 303 | isoform X1: XP_016865549.1, 016865550.1, and XP_016865551.1 are identical; alternative splice | 32-HKSRSTSRGDVNDQDAIQEKF-52 |
| XP_016865550.1 | 303 | isoform X1: XP_016865549.1, 016865550.1, and XP_016865551.1 are identical; alternative splice | 32-HKSRSTSRGDVNDQDAIQEKF-52 |
| XP_016865551.1 | 303 | isoform X1: XP_016865549.1, 016865550.1, and XP_016865551.1 are identical; alternative splice | 32-HKSRSTSRGDVNDQDAIQEKF-52 |
achr14:37,818,807 A/G is the position of the equine V238A variant of MYOT as displayed in the UCSC Browser. Please note that this corresponds to V237A in NP_006781.1.
bAccession ID at NCBI; click Accession ID in table to go to sequence.
cNumber of amino acids (aa) in predicted protein
dXP_014586147 is the full-length horse MYOT protein sequence. All others are human MYOT protein sequences that include the site of the equine V238A variant. See text for details.
eThe amino acid sequence of the indicated protein is shown around the site of the amino acid affected by the equine V238A variant, which is indicated in bold and underlining. The numbers flanking the sequence indicate the positions of the first and last amino acids in the 21 amino acid sequence shown.
Table 4 compares the sequence of the equine MYOT protein to the sequence of the human MYOT protein around the site of the equine S233P variant. Eight human protein sequence IDs, representing four different protein sequences, are compared following sequence alignment. The sequence of the four different human sequences represented by eight entries at NCBI are identical at the variant site. The sequences of the ten amino acids N-terminal to the variant site are identical in isoforms a, b, and c, and are also identical to the equine sequence, but differ from the sequence in isoform X1. The sequences of the ten amino acids C-terminal to the variant site are identical among all isoforms and are identical to the equine sequence.
All human protein sequences are identical at the variant site, and differ from the horse protein sequence by having a threonine (T) rather than a serine (S) at this position, a conservative substitution. The substitution of a threonine (T) for a serine (S) at this position is a feature of the human protein and also of proteins of primates closely related to humans, but other mammals have a serine (S) at this position as seen in horse. The equine S233P variant is a nonconservative substitution of a proline (P) for a serine (S) at this position, and is expected to be damaging. Please see the patent cited by the authors (patent #WO2017165733A1) for more details.
Therefore, even when the previously noted error assigning the V238A variant to the position of the S233P variant (chr14:37,818,823 A/G) is corrected, there is no support for the author’s statement that the amino acid at the position of the S233P equine variant differs among human isoforms.
| Table 4. Human isoforms of MYOT compared to horse MYOT near chr14:37,818,823 A/Ga | |||
|---|---|---|---|
| Accession IDb | Length (aa)c | Notesd | Sequence around equine S233P variante |
| XP_014586147.1 | 498 | Full length horse protein sequence | 222-VPTSQVRSRSSSRGDVNDQDA-242 |
| Q9UBF9.2 | 498 | isoform a: NP_006781.1, BAG35604.1, and Q9UBF9.2 are identical | 222-VPTSQVRSRSTSRGDVNDQDA-242 |
| BAG35604.1 | 498 | isoform a: NP_006781.1, BAG35604.1, and Q9UBF9.2 are identical | 222-VPTSQVRSRSTSRGDVNDQDA-242 |
| NP_006781.1 | 498 | isoform a: NP_006781.1, BAG35604.1, and Q9UBF9.2 are identical | 222-VPTSQVRSRSTSRGDVNDQDA-242 |
| NP_001287840.1 | 383 | isoform c: N-terminal 115 aa sequence missing vs. NP_006781.1 | 107-VPTSQVRSRSTSRGDVNDQDA-127 |
| NP_001129412.1 | 314 | isoform b: N-terminal 184 aa sequence missing vs. NP_006781.1 | 38-VPTSQVRSRSTSRGDVNDQDA-58 |
| XP_016865549.1 | 303 | isoform X1: XP_016865549.1, 016865550.1, and XP_016865551.1 are identical; alternative splice | 27-CKFLHHKSRSTSRGDVNDQDA-47 |
| XP_016865550.1 | 303 | isoform X1: XP_016865549.1, 016865550.1, and XP_016865551.1 are identical; alternative splice | 27-CKFLHHKSRSTSRGDVNDQDA-47 |
| XP_016865551.1 | 303 | isoform X1: XP_016865549.1, 016865550.1, and XP_016865551.1 are identical; alternative splice | 27-CKFLHHKSRSTSRGDVNDQDA-47 |
achr14:37,818,823 A/G is the position of the equine S233P variant of MYOT as displayed in the UCSC Browser. Please note that this corresponds to S232P in NP_006781.1.
bAccession ID at NCBI; click Accession ID in table to go to sequence.
cNumber of amino acids (aa) in predicted protein
dXP_014586147 is the full-length horse MYOT protein sequence. All others are human MYOT protein sequences that include the site of the equine S233P variant. See text for details.
eThe amino acid sequence of the indicated protein is shown around the site of the amino acid affected by the equine S233P variant, which is indicated in bold and underlining. The numbers flanking the sequence indicate the positions of the first and last amino acids in the 21 amino acid sequence shown.
REQUEST FOR PRINTED CORRECTION: Unless the authors can provide evidence of the kind presented above for their statement that the amino acid affected by the MYOT genetic variant (chr14:37,818,823 A/G) analyzed in the patent that they cite (patent #WO2017165733A1) differs among human isoforms of the MYOT protein, we request that the Equine Veterinary Journal print a correction to this article [1].
Conclusions
This study screens a limited number of candidate genes in horses whose human orthologs are associated with muscle disease to identify 26 missense alleles in these genes. A set of eight apparently unrelated Warmblood horses with MFM as diagnosed by desmin staining of muscle samples is compared to a set of eight control Warmbloods. No statistically significant difference is seen between the two sets of samples for any of the variants.
In the discussion [1], the authors state:
To date, the genetic myopathies identified in horses have been monogenic and given equine founder effects and the structure of horse breed types, it is reasonable to hypothesize that horses sharing disease pathology also share the same causative variant. Polygenic inheritance is certainly possible, however, inspection of the heat map generated for variants in 16 MFM candidate genes in all horses did not identify a consistent pattern in which a set of multiple variants were present in MFM WB and not present in controls.
It is not surprising that genetic myopathies identified in horses to date have been monogenic. There are only a small number of equine inherited disorders of any kind currently associated with specific genetic defects. In such a small collection of disorders with an identified genetic cause, it is to be expected that monogenic disorders, which are the easiest to find, make up the entire sample.
Horses with a diagnosis of PSSM2 or MFM as assigned by results of muscle biopsy have been analyzed using GWAS by another research group. If the disease were monogenic, this approach would have been successful at identifying a candidate region. GWAS was successful at identifying the GYS1-R309H variant associated with Polysaccharide Storage Myopathy type 1 (PSSM1), one of the monogenic disorders referred to by the authors [22].
If Warmblood MFM, as diagnosed by a positive desmin stain, were monogenic and associated with a genetic variant in any of the sixteen genes analyzed in this study, it should have been possible to identify the causative genetic variant even in a study of this size (eight affected and eight control horses).
The authors dismiss a hypothesis of polygenic inheritance in which they expect variants of the sixteen genes analyzed to display a statistically significant difference in frequency between the affected and control Warmbloods. A study of this size applied to unrelated human patients with MFM would likely not have been able to demonstrate a statistically significant association with variants of any of the nine genes currently associated with MFM in human patients.
This study therefore is only successful at refuting the hypothesis that Warmblood MFM is a monogenic disorder associated with genetic variants in any of the sixteen genes analyzed. With respect to the possibility of polygenic inheritance, the results of this statistically underpowered trial are at best inconclusive.
References
[1] Williams ZJ, Velez-Irizarry D, Petersen JL, Ochala J, Finno CJ, and Valberg SJ (2020) Candidate gene expression and coding sequence variants in Warmblood horses with myofibrillar myopathy. Equine Vet J. 00: 1-10. PMID: 32453872
[2] McLaren W et al. (2016) The Ensembl Variant Effect Predictor. Genome Biol 17(1): 122. PMID: 27268795
[3] Goldfarb LG et al. (1998) Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nature Genet. 19: 402-403. PMID: 9697706
[4] Valberg SJ et al. (2016) Suspected Myofibrillar Myopathy in Arabian horses With a history of exertional rhabdomyolysis. Equine Vet J. 48(5): 548-556. PMID: 26234161
[5] Valberg SJ et al. (2017) Clinical and histopathological features of Myofibrillar Myopathy in Warmblood horses. Equine Vet J. 49(6): 739-745. PMID: 28543538
[6] Pfeffer G et al. (2014) Titin founder mutation is a common cause of myofibrillar myopathy with early respiratory failure. J. Neurol. Neurosurg. Psychiat. 85: 331-338. PMID: 23486992
[7] Gundesli H et al. (2010) Mutation in exon 1f of PLEC, leading to disruption of plectin isoform 1f, causes autosomal-recessive limb-girdle muscular dystrophy. Am. J. Hum. Genet. 87: 834-841. PMID: 21109228
[8] Smith FJD et al. (1996) Plectin deficiency results in muscular dystrophy with epidermolysis bullosa. Nature Genet. 13: 450-457. PMID: 8696340
[9] Pulkkinen L et al. (1996) Homozygous deletion mutations in the plectin gene (PLEC1) in patients with epidermolysis bullosa simplex associated with late-onset muscular dystrophy. Hum. Molec. Genet. 5: 1539-1546. PMID: 8894687
[10] Koss-Harnes D et al. (2002) A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: two identical de novo mutations. J. Invest. Derm. 118: 87-93. PMID: 11851880
[11] Pfendner E and Uitto J (2005) Plectin gene mutations can cause epidermolysis bullosa with pyloric atresia. J. Invest. Derm. 124: 111-115. PMID: 15654962
[12] Gostynska KB et al. (2015) Mutation in exon 1a of PLEC, leading to disruption of plectin isoform 1a, causes autosomal-recessive skin-only epidermolysis bullosa simplex. Hum. Molec. Genet. 24: 3155-3162. PMID: 25712130
[13] Bucelli RC et al. (2015) SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 85: 665-674. PMID: 26208961
[14] Fecto F et al. (2011) SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 68: 1440-1446. PMID: 22084127
[15] Rubino E et al. (2012) SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 79: 1556-1562. PMID: 22972638
[16] Hirano M et al. (2013) Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology. 80: 458-463. PMID: 23303844
[17] Le Ber I et al. (2013) SQSTM1 mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 70: 1403-1410. PMID: 24042580
[18] Boutoleau-Bretonniere C et al. (2015) A phenotype of atypical apraxia of speech in a family carrying SQSTM1 mutation. J. Alzheimers Dis. 43: 625-630. PMID: 25114083
[19] Haack T. B. et al. (2016) Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with ataxia, dystonia, and gaze palsy. Am. J. Hum. Genet. 99: 735-743. PMID: 27545679
[20] Laurin N et al. (2002) Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am. J. Hum. Genet. 70: 1582-1588. PMID: 11992264
[21] Hocking LJ et al. (2002) Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum. Molec. Genet. 11: 2735-2739. PMID: 12374763
[22] McCue M et al. (2008) Glycogen Synthase (GYS1) Mutation Causes a Novel Skeletal Muscle Glycogenosis. Genomics. 91(5): 458-466. PMID: 18358695
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
P4 Allele of MYOZ3
Introduction This blog post explores the P4 allele (S42L) of the equine MYOZ3…
1 Apr 2025
P2 Allele of MYOT
Introduction This blog post explores the P2 allele (S232P) of the equine MYOT…
8 Mar 2025
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
© 2017 – 2023 EquiSeq Inc. All rights reserved.