Study of horse genomes explores genetic burden
What is the genetic load in horses?
What are loss-of-function variants?
Stop gain
Figure 1
Stop loss
Figure 2
Start loss
Figure 3
Frameshift
Figure 4
Splice site alteration
Figure 5
Missense
Figure 6
Sources of error in the identification of loss-of-function variants
Loss-of-function alleles present at high frequencies
Strong phenotypes
Table 1
Unknown or mild phenotypes
Table 2
Poor transcript models
Table 3
Figure 7
Additional annotation errors
Table 4
Summary
Table 5
What does allele frequency tell us?
Allele frequency of disease-associated variants
Table 6
Evaluation of missense alleles
Introduction
A team of researchers at the University of Minnesota and the University of California, Davis, have published a landmark study of the predicted genetic burden in horses, based on the analysis of whole genome sequence data from 605 horses (1). They conclude that the genetic load in horses is 1.4 – 2.6 times that of the human population. The authors discuss the unique advantages of the study of horse to understand human phenotypes, especially those associated with athletic performance.
Horse owners have asked us to explain this paper, as it mentions the genetic variants that are in EquiSeq’s panel of DNA tests. Here we review the methods and major findings of this paper. We include background information typically absent from the primary literature in order to make the paper more accessible to non-specialists.
This is a long post and is necessarily technical. If you are looking for a fast answer, please skip to the Conclusions.
What is genetic load?
Genetic load (also called genetic burden) is a concept from population genetics. It assumes that there is an optimal genotype free of all harmful genetic variants for that species. Genetic load is the difference in fitness between the average individual in that population and the “perfect” individual with the optimal genotype. For a further discussion of genetic load, please see recent reviews (2, 3, 4, 5).
What is the genetic load in horses?
In this study, the authors use two computational variant impact predictors to analyze single-nucleotide polymorphisms (SNPs) and small insertions and deletions found in the 605 horse genomes. Variants considered high impact by both programs, or high impact by one program and moderate impact by the other, were classified as high impact variants and were used to estimate genetic load. This resulted in a list of 25,944 high impact variants affecting 9,387 genes.
What are loss-of-function variants?
Mutations in the horse genome may be classified by type. The most severe mutations are expected to cause a complete loss of function of the encoded protein. Loss-of-function variants include:
1. Stop gain: A stop gain or nonsense allele has a mutation that changes one of the amino-acid-encoding codons to a stop codon. This will result in the synthesis of a truncated protein. Almost all nonsense alleles where the mutation is not located in the terminal part of the coding sequence will be loss-of-function alleles.

Figure 1. Stop gain (nonsense) allele. Mutation of a single base in the TCG (serine) codon changes it to a TAG (termination or stop) codon.
2. Stop loss: The last codon of the coding sequence is one of three stop codons that cause protein synthesis to stop at that position. A mutation that causes the loss of the stop codon will result in a protein with a carboxy-terminal extension whose length is specified by the next stop codon to be reached. It is possible that this will not cause a complete loss of function for some genes.

Figure 2. Stop loss allele. Mutation of a single base in the TAA (stop) codon changes it to a TAC (tyrosine) codon.
3. Start loss: The first codon of the coding sequence of most genes is ATG (encoding methionine). This codon is recognized by the cell’s translational machinery as the starting point for protein synthesis. Loss of the start codon may result in the complete absence of protein expression, or the translational machinery may initiate protein synthesis at another site, resulting in a partial protein or a protein with a frameshift.

Figure 3. Start loss allele. Mutation of a single base in the ATG (start/methionine) codon changes it to a TTG (leucine) codon that is incapable of serving as the position for initiation of protein synthesis.
4. Frameshift: An insertion or deletion of a number of bases in a coding region will alter the reading frame if the number of bases is not a multiple of three. When the transcript containing a frameshift mutation is translated, the amino acid sequence of the encoded protein will be altered downstream of the frameshift variant. The out-of-frame sequence is likely to contain a stop codon causing premature termination of the protein sequence. Almost all frameshift alleles except for those in the very terminal part of the coding sequence are expected to cause a loss of protein function.

Figure 4. Frameshift allele. Deletion of a single base in the first codon alters that codon from AGT (serine) to AGG (arginine); all downstream codons are read in the wrong reading frame, resulting the alteration of protein sequence.
5. Splice site alteration: Mature eukaryotic mRNA is not an identical copy of the region of the genome containing a gene. The primary transcript is spliced, with coding sequence (exons) conserved, and sequences that interrupt the coding sequence (introns) removed. The boundaries between introns and exons contain short sequences that specify the removal of an intron to the splicing machinery. Mutation of these sites will alter the splicing of the primary transcript, resulting in a mature mRNA that has one or more exons missing, potentially causing a frameshift downstream of the mutation. Splice site alterations are therefore recognized as loss-of-function variants.
The primary transcripts of many genes show alternative splicing, which allows a single gene to make a family of related proteins (isoforms). A splice site alteration that affects an exon that is not common to all isoforms might not cause a complete loss of function.
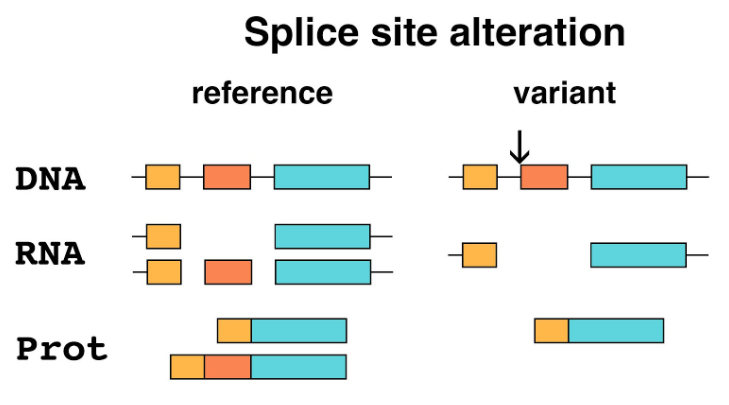
Figure 5. Splice site alteration. This example shows DNA that is part of a gene with three exons (color) separated by introns (thin lines). The reference allele in this example produces two different isoforms by alternative splicing, one including the middle exon and one excluding it. The variant (arrow) eliminates the splice site at the beginning of the second exon. Only one protein isoform lacking the middle exon is produced.
All five of these types of mutations are reasonably considered to cause loss of protein function in most cases. Only variants classified as loss-of-function variants by both programs were considered further, resulting in a catalog of 18,990 loss-of-function variants representing 7,682 genes in the 605 horse genomes (1).
There is one additional type of mutation that is not part of the bulk analysis of genomes in this paper.
6. Missense: Alteration of a single base within the coding region of a gene can either result in a synonymous substitution (not considered further here), or a nonsynonymous substitution of a different amino acid (a missense allele). The authors of the current study (1) do not analyze missense alleles in their bulk analysis, but comment on the allele frequency of some disease-associated missense alleles.

Figure 6. Missense allele. Mutation of a single base in the coding region changes a CGT (arginine) codon to a CAT (histidine) codon.
The authors exclude all missense alleles from consideration as loss-of-function alleles. Many missense alleles are known to be benign, with no effects on protein function or phenotype. Others are known to be loss-of-function variants. Still others have minor effects on protein function or phenotype.
Kryukov et al. (6) analyzed missense alleles in the human population and estimated that ~20% result in loss of function, ~27% are effectively neutral, and that ~53% of new missense alleles are mildly deleterious. The evaluation of missense alleles is more challenging than the evaluation of loss-of-function alleles of the other five types described above. In the discussion near the end of this blog, we discuss the evaluation of missense alleles using allele frequency (a method used by the authors), the chemistry of the amino acid substitution (not considered by the authors), and evolutionary conservation of the affected amino acid (not considered by the authors).
Estimate of genetic burden
The authors use their catalog of variants to examine genetic load in 493 horses representing twelve breeds with whole genome sequence data from 17 or more individuals. The results show a predicted genetic burden in horse that is approximately twice that of the human population. Predicted genetic burden (all variants) and loss-of-function variants for the average horse are 730 and 417, respectively, while comparable estimates for the human population are 281-515 genetic burden variants and 250-300 loss-of-function variants per individual.
The higher genetic burden seen in horse may result from population bottlenecks or selection for particular traits. It is also possible that the higher genetic burden seen in horse is somewhat inflated by a number of factors identified in a similar analysis of human genomes (7).
Sources of error in the identification of loss-of-function variants
MacArthur et al (7) analyzed 2,951 putative loss-of-function alleles from 185 human genomes and estimated that the average human genome has 100 loss-of-function variants and that 20 of these are homozygous, completely eliminating gene function. This study took particular care in characterizing loss-of-function variants, and identified the following sources of error:
1. Sequencing and mapping errors: Short-read DNA sequencing produces a set of sequences that are mapped to a model of the genome (the reference assembly). There are two possible sources of error in this process.
DNA sequencing errors. DNA sequencing produces a low frequency of errors, so in whole genome sequencing, each part of the genome is sequenced multiple times. A high quality whole genome sequence might have 30x coverage, while whole-genome sequencing done to survey a large number of individuals for variants might have 4x – 6x coverage.
Mapping error. In some cases, sequences derived from a non-functioning duplicate copy of a gene (a pseudogene) are incorrectly mapped to the functional copy and are incorrectly scored as loss-of-function variants of the gene. In other cases, the depth of short-read coverage is insufficient for an accurate mapping to the assembly; this is likely when the sequence in question contains repeated sequences, even if these are not identical.
2. Reference sequence errors: The reference assembly of a genome is a model that is periodically revised. In some cases, the sequence of a gene has minor errors (assembly errors) that would make neutral genetic variants appear to be loss-of-function variants.
3. Partial loss-of-function variants: The automated identification of candidate loss-of-function variants identifies some that are unlikely to be true loss-of-function variants. These include nonsense and frameshift mutations near the terminus of the coding region, stop-loss mutations that produce only a small terminal extension, or any kind of mutation including splice site alterations that affect an exon that is not present in all isoforms.
In the study by MacArthur et al. (7), 25% of candidate loss-of-function variants were eliminated as sequencing and mapping errors, 27% of candidate loss-of-function variants were eliminated as annotation or reference sequence errors, and 11% of candidate loss-of-function variants were eliminated as unlikely to be genuine loss-of-function variants. Only 43.5% of the candidate loss-of-function alleles survived filtering for the three sources of error cited above.This raises the possibility that in the study under discussion here (1), the number of loss-of-function variants identified in 605 horse genomes is inflated by a lower quality reference assembly for the horse genome and a lack of filtering of candidate loss-of-function variants to identify those that are partial loss-of-function variants. It is difficult to compare this study directly with the work of MacArthur et al. (7), because improvements to human genome annotation and to the human genome assembly since then have likely improved the quality of computationally-derived gene models in horse.
Loss-of-function alleles present at high frequencies
Putative loss-of-function alleles that are present at high frequencies in the general horse population might identify genes for which loss-of-function variants produce no readily observable phenotype. It is also possible that the analysis presented in this study (1) has identified assembly errors in the reference horse genome that will require manual curation to resolve.
Supplementary Table 6 (1) gives Ensembl IDs for all genes with loss-of-function variants present at allele frequencies higher than 5%. There are 725 such genes, 7.7% of the 9,387 genes identified as carrying loss-of-function variants. Some of these genes are associated with multiple loss-of-function variants, but we do not consider this further in this review.
MacArthur et al (7), screening the human genome for loss-of-function variants, manually reannotated genes with loss-of-function alleles at high frequencies. The authors of the current study do not describe such work, so we have examined 100 Ensembl IDs with the highest frequency of loss-of-function alleles from Supplementary Table 6.
Ensembl IDs from Supplementary Table 6 were used to search the horse genome in the UCSC Genome Browser to examine the quality of annotation. Some Ensembl IDs are not associated with transcript models from human or mouse. In other cases, Ensembl IDs align to transcript models from human, mouse, and other species. In some cases, it is clear that the computational annotation is not accurate and that manual reannotation is required. We present a summary of the 100 apparent loss-of-function variants with the highest allele frequencies, evaluated using publicly available data from UniProt, OMIM, and Mouse Genome Informatics (MGI).
There are 11 olfactory receptor genes in the list of 100 genes. These genes belong to a large, rapidly evolving gene family and are not considered further here.
Information on human phenotypes comes from clinical studies on rare diseases, often identified in consanguineous pedigrees. Information on mouse phenotypes generally comes from knock-out alleles that have been engineered to eliminate gene function. This means that phenotype data from mouse represents loss-of-function phenotype more accurately than does phenotype data from human populations.
Strong phenotypes
There are 18 genes for which loss-of-function variants in human or mouse produce an obvious phenotype that likely would have been identified in horse. With the exception of ASIP, a common variant of which causes nonagouti coat color, it is unlikely that any of these variants have been called correctly, and manual re-annotation is called for in each case. Table 1 summarizes genes in this category.
Table 1. Loss-of-function variants with obvious phenotypes in human or mouse. The Human column gives links to the disease entry in OMIM. The Mouse column gives a summary of knockout phenotypes from Mouse Genome Informatics, highlighting phenotypes that would be most evident in horse.
| Ensembl ID | Gene | Human | Mouse |
| ENSECAG00000013126 | WFS1 | 222300 | deafness, impaired glucose tolerance |
| ENSECAG00000016526 | NUBPL | 618242 | embryonic/perinatal lethal |
| ENSECAG00000012463 | RPS20 | N/A | embryonic/perinatal lethal |
| ENSECAG00000018661 | EXTL3 | 617425 | embryonic/perinatal lethal |
| ENSECAG00000016969 | STRA6 | 601186 | N/A |
| ENSECAG00000021649 | ZNF266 | N/A | perinatal lethality |
| ENSECAG00000010440 | SYF2 | N/A | embryonic lethality |
| ENSECAG00000019625 | HGSNAT | 252930 | behavior, internal anatomy |
| ENSECAG00000021316 | FAM21A | N/A | morphology |
| ENSECAG00000015945 | SLC17A5 | 269920 | abnormal gait |
| ENSECAG00000025038 | LOXL2 | N/A | perinatal lethality |
| ENSECAG00000013708 | SLC24A4 | 615887 | amelogenesis imperfecta |
| ENSECAG00000035776 | TAC3 | 614839 | reproductive system anomalies |
| ENSECAG00000014440 | DISP1 | N/A | embryonic lethality |
| ENSECAG00000037215 | RFK | N/A | embryonic/perinatal lethality |
| ENSECAG00000010236 | XPA | 278700 | UV sensitivity |
| ENSECAG00000007192 | PTPRC | 619924 | immunodeficiency |
| ENSECAG00000004241 | ASIP | 611742 | nonagouti |
Unknown or mild phenotypes
There are 12 genes for which there are no data on loss-of-function variants in humans or mice. There are 26 genes with phenotype data from human or mouse that suggest that loss-of-function variants would not be immediately apparent in horse. Table 2 summarizes genes in this category.
Table 2. Loss-of-function variants with no information on phenotypes in human or mouse, or phenotypes in human or mouse that might have been overlooked in horse. The Human column gives links to the disease entry in OMIM. The Mouse column gives a summary of knockout phenotypes from Mouse Genome Informatics, highlighting phenotypes that would be most evident in horse.
| Ensembl ID | Gene | Human | Mouse |
| ENSECAG00000015417 | RBM11 | N/A | N/A |
| ENSECAG00000000927 | IFT70B | N/A | internal anatomy |
| ENSECAG00000013289 | IFNLR1 | N/A | susceptibility to viral infection |
| ENSECAG00000014769 | ITPKB | N/A | sparse fur, T cell alterations |
| ENSECAG00000024895 | PRELID2 | N/A | internal anatomy |
| ENSECAG00000012110 | MRPL15 | N/A | N/A |
| ENSECAG00000014006 | MLN | N/A | N/A |
| ENSECAG00000010758 | KIF24 | N/A | N/A |
| ENSECAG00000007511 | SPOCK1 | N/A | decreased blood glucose, grip strength |
| ENSECAG00000007936 | SARDH | 268900 | body weight, abnormal spatial memory |
| ENSECAG00000014784 | FCMR | N/A | alterations to immune system |
| ENSECAG00000022993 | MTMR1 | N/A | increased body fat |
| ENSECAG00000010626 | SLC47A2 | N/A | decreased bone mineral density |
| ENSECAG00000036676 | GGACT | N/A | NA |
| ENSECAG00000024146 | UCMA | N/A | no phenotype |
| ENSECAG00000039152 | GABARAPL1 | N/A | N/A |
| ENSECAG00000022609 | CHRNA2 | 610353 | normal behavior |
| ENSECAG00000020682 | TTLL11 | N/A | N/A |
| ENSECAG00000016989 | OTOL1 | N/A | internal anatomy |
| ENSECAG00000017519 | PDE9A | N/A | decreased cardiac response to stress |
| ENSECAG00000021344 | ST6GALNAC2 | N/A | decreased body weight |
| ENSECAG00000033923 | RHOQ | N/A | behavior, brain anatomy |
| ENSECAG00000033857 | PILRA | N/A | internal anatomy, blood chemistry |
| ENSECAG00000016521 | PTGIS | 145500 | renal fibrosis, internal anatomy |
| ENSECAG00000008850 | OSCP1 | N/A | internal anatomy, hematocrit |
| ENSECAG00000011079 | MRPL19 | N/A | N/A |
| ENSECAG00000005649 | PAQR7 | N/A | N/A |
| ENSECAG00000021196 | ACOT12 | N/A | liver fibrosis, blood chemistry |
| ENSECAG00000025154 | ADPRM | N/A | behavior |
| ENSECAG00000014585 | PLAC8 | N/A | N/A |
| ENSECAG00000017907 | RNFT2 | N/A | internal anatomy |
| ENSECAG00000022297 | TEX45 | N/A | no phenotype |
| ENSECAG00000016814 | PPM1A | N/A | bone structure, abnormal wound healing |
| ENSECAG00000017087 | ACSBG2 | N/A | decreased bone mineral density |
| ENSECAG00000006195 | FPR2 | N/A | internal anatomy, immune respones |
| ENSECAG00000005924 | TSKU | N/A | internal anatomy |
| ENSECAG00000036594 | NUDT3 | N/A | N/A |
| ENSECAG00000015003 | PLGRKT | N/A | dermatitis |
Poor transcript models
Ensembl IDs associated with proposed loss-of-function variants are computational models of transcripts. Using the reported Ensembl IDs to search the horse genome in the UCSC Genome Browser reveals horse transcript models that fail to align with transcript models from human or mouse. In some cases, the Ensembl model fails to match any known gene. In other cases, the model differs substantially from transcript models for human and mouse.
There are 28 variants for which the Ensembl transcript model used to identify the variant does not align to transcripts from human or mouse. Most of these cases probably do not identify actual horse genes. There are five variants for which the Ensembl transcript model used to identify the variant partially aligns to transcripts from human or mouse, but in the cases identified in Table 3, there is an obvious annotation error. There are annotation errors apparent in the genes listed in Table 1 and Table 2; these are listed in Table 4.
Table 3. Loss-of-function variants likely resulting from annotation errors.
| Ensembl ID | Gene | Alignment to human or mouse transcripts |
| ENSECAG00000032170 | ? | none |
| ENSECAG00000034616 | EIF3J1 | poor |
| ENSECAG00000035736 | ? | none |
| ENSECAG00000031545 | ? | none |
| ENSECAG00000028106 | ? | none |
| ENSECAG00000036544 | ? | none |
| ENSECAG00000041059 | ? | none |
| ENSECAG00000036901 | ? | none |
| ENSECAG00000032890 | ? | none |
| ENSECAG00000038480 | ? | none |
| ENSECAG00000036718 | ? | none |
| ENSECAG00000035289 | ? | none |
| ENSECAG00000034443 | ? | none |
| ENSECAG00000035877 | ? | none |
| ENSECAG00000042911 | LOC102149855 | poor |
| ENSECAG00000031568 | ? | none |
| ENSECAG00000032770 | ? | none |
| ENSECAG00000043325 | ? | none |
| ENSECAG00000020682 | LOC111767962 | poor |
| ENSECAG00000043476 | ? | none |
| ENSECAG00000037799 | ? | none |
| ENSECAG00000016698 | TSBP1 | poor |
| ENSECAG00000013903 | ? | none |
| ENSECAG00000039092 | ? | none |
| ENSECAG00000029891 | ? | none |
| ENSECAG00000031548 | ? | none |
| ENSECAG00000003430 | ? | none |
| ENSECAG00000011190 | METM/ANTKMT fusion | good |
| ENSECAG00000038600 | ? | none |
| ENSECAG00000035012 | ? | none |
| ENSECAG00000007603 | ? | none |
| ENSECAG00000036375 | ? | none |
| ENSECAG00000030484 | ? | none |
One good example of an annotation error is the METM/ANTKMT fusion, shown in the figure below. The Ensembl transcript model used to identify a loss-of-function allele from Supplementary Table 6 predicts a 3’ exon not seen in any other species.
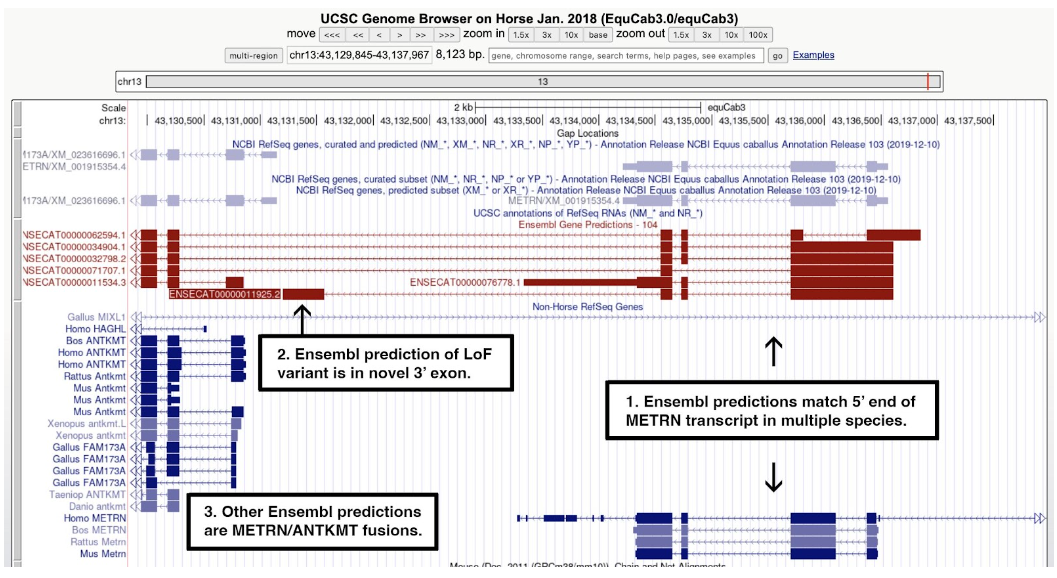
Figure 7. A screenshot of the UCSC Genome Browser centered on ENSECAG00000011190. Text added to the figure shows that: 1) multiple Ensembl transcript models (red) match the 5’ end of transcripts of METRN in human, mouse, rat, and cattle (blue); 2) the search on the selected Ensembl ID from the list of loss-of-function alleles highlighted a model with a novel 3’ exon not seen in any other transcript model; and 3) other Ensembl models fuse METM to ANTKMT. The RefSeq model (top right) matches one of the Ensembl models and aligns well to METM orthologs from human, mouse, rat, and cattle.
Additional annotation errors
Table 4. Likely annotation errors from Tables 1 and 2.
| Ensembl ID | Gene | Table | Error |
| ENSECAG00000000927 | IFT70B | 2 | Model misses intron seen in multiple species |
| ENSECAG00000013126 | WFS1 | 1 | 5’ exon doesn’t align to other species, AGG start |
| ENSECAG00000014769 | ITPKB | 2 | novel exon doesn’t align to other species |
| ENSECAG00000024895 | PRELID2 | 2 | novel 3’ exon doesn’t align to other species |
| ENSECAG00000012463 | RPS20 | 1 | novel 3’ exon doesn’t align to other species |
| ENSECAG00000018661 | EXTL3 | 1 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000016969 | STRA6 | 1 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000014784 | FCMR | 2 | novel 3’ exon doesn’t align to other species |
| ENSECAG00000022993 | MTMR1 | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000010626 | SLC47A2 | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000036676 | GGACT | 2 | novel 3’ exon doesn’t align to other species |
| ENSECAG00000039152 | GABARAPL1 | 2 | novel 5’ exon, incomplete gene model |
| ENSECAG00000016989 | OTOL1 | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000021344 | ST6GALNAC2 | 2 | aligns poorly to other species |
| ENSECAG00000033923 | RHOQ | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000033857 | PILRA | 2 | novel 5’ exons don’t align to other species |
| ENSECAG00000016521 | PTGIS | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000025038 | LOXL2 | 1 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000035776 | TAC3 | 1 | novel 3’ exon doesn’t align to other species |
| ENSECAG00000011079 | MRPL19 | 2 | missing 3’ exons, transcript extended |
| ENSECAG00000025154 | ADPRM | 2 | missing 3’ exons, transcript extended |
| ENSECAG00000014585 | PLAC8 | 2 | missing 5’ exon, transcript extended |
| ENSECAG00000025038 | LOXL2 | 1 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000035776 | TAC3 | 1 | 3’ exon aligns poorly to other species |
| ENSECAG00000011079 | MRPL19 | 2 | missing 3’ exon, transcript extended |
| ENSECAG00000025154 | ADPRM | 2 | missing 3’ exon, transcript extended |
| ENSECAG00000016814 | PPM1A | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000017087 | ACSBG2 | 2 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000037215 | RFK | 1 | novel 5’ exon doesn’t align to other species |
| ENSECAG00000005924 | TSKU | 2 | novel 3’ exon doesn’t align to other species |
| ENSECAG00000036594 | NUDT3 | 2 | aligns poorly to other species |
Summary
Our classifications of the 100 most frequent putative loss-of-function alleles from Supplementary Table 6 are shown in Table 5.
Table 5. Classification of 100 loss-of-function alleles from Supplementary Table 6
| Type | Observed |
| Olfactory | 11 |
| Strong phenotype, benign | 1 |
| Strong phenotype | 17 |
| Mild phenotype | 27 |
| No phenotype | 2 |
| Unknown phenotype | 10 |
| Poor transcript model | 32 |
| Total | 100 |
Of the 89 genes that are not olfactory receptors, 17 are removed from further consideration as loss-of-function alleles in horse because they are expected to produce strong phenotypes, and 32 are removed from further consideration because they are based on poor transcript models. This leaves 40 genes expected to produce a mild phenotype, an unknown phenotype, or no phenotype. The only loss-of-function variant on this list that does not require additional analysis is ASIP, responsible for nonagouti coat color.
The loss-of-function variants present at the highest allele frequencies are expected to be enriched for annotation errors and genes tolerant to loss-of-function alleles, so elimination of the majority of the loss-of-function variants from further consideration in this list does not generalize to the remainder of the 9,387 genes identified as having loss-of-function alleles in this study.
What does allele frequency tell us?
In the well-studied human population, loss-of-function variants are generally present at allele frequencies below 5%, presumably due to purifying selection against these variants. This has proven to be a rule of general utility in the evaluation of large numbers of variants in the human genome, but it needs to be applied with caution.
There is evidence here that the identification of loss-of-function variants in horses is mostly accurate. The median allele frequency of loss-of-function variants in the 605 horses in this study is 0.16% (1), consistent with selection against loss-of-function variants in most genes, as seen in human populations (7).
Yet some loss-of-function alleles are present in the human population at frequencies greater than 50% (7). This is interpreted to mean that the human (and presumably the horse) genome is somewhat buffered against loss-of-function alleles, with some genes performing functions that are redundant or dispensable.
This study was blind with respect to phenotype; there is no phenotypic information on the 605 horses analyzed overall or on the 493 horses representing twelve breeds. The authors document the allele frequency of disease-associated alleles found in OMIA. Some of these damaging variants are present in frequencies above 5% in some breeds or subtypes.
The highest possible allele frequency for a recessive variant that has a strong phenotype preventing homozygous individuals from reproducing is 50%. Imagine a population in which every individual is heterozygous for a recessive lethal mutation, and there are no individuals that are homozygous for the wild-type allele. In the next generation, the 1:2:1 ratio of genotypes will become a 1:2 ratio of homozygous wild type to heterozygotes for the lethal. This will cause the allele frequency to drift downward due to purifying selection.
Therefore, variants identified as a loss-of-function alleles predicted to have a recessive lethal phenotype cannot have an allele frequency in excess of 50%; any cases above this frequency cannot be a correct call. For example, ENSECAG00000013126 identifies a putative loss-of-function allele of WFS1, which in human patients is associated with diabetes, deafness, and optic atrophy. The putative loss-of-function allele is present at a frequency of 78%.
In some cases, however, there is balancing selection. This means that both the homozygotes for a recessive lethal variant and homozygotes for the reference allele are selected against. In pigs, a 212 kb deletion acts both as a recessive lethal and as a dominant variant that increases weight gain (8). The allele frequency of the deletion is 5.4%.
In chickens, a missense allele of OFD1 appears to be associated with recessive lethality despite an allele frequency of 8.9% (9). In this case, it is not clear whether the relatively high allele frequency is due to balancing selection or drift. Splice variants for CHTF18 and FLT4 show allele frequencies higher than 5% (9).
Allele frequency of disease-associated variants
Compared to the human population, a relatively small number of genetic variants are currently associated with inherited disease in horse. We briefly discuss examples raised by the authors as well as other examples from the peer-reviewed literature.
PKHD1: The PKHD1 gene encodes a signaling receptor involved in morphogenesis of the cilium and centrosome as well as the orientation of the mitotic spindle. In human patients, mutations in this gene are associated with Polycystic Kidney Disease. Two publications report an association of PKHD1 with Congenital Liver Fibrosis in horses (10, 11), but in this study, one of the PKHD1 variants is seen at an allele frequency of 89.5% in Clydesdales, and homozygotes have been seen in other breeds (1).
The allele frequency reported in this study effectively eliminates the PKHD1 variants as candidates for the cause of Congenital Liver Fibrosis.
TRIM1-RPP2: TRIM1 and RPP2 are adjacent genes affected by a deletion that was initially associated with Juvenile Idiopathic Epilepsy in horses (12), although this association did not hold up in other studies (13, 14). In this study, the deletion was found to be the major allele in Standardbreds and Arabians (1).
The allele frequency reported in this study and prior studies effectively eliminates the TRIM1-RPP2 deletion as a candidate for the cause of Juvenile Idiopathic Epilepsy.
SCN4A: The SCN4A gene encodes a pore-forming subunit of a voltage-gated sodium channel complex. A dominant variant allele is associated with Hyperkalemic Periodic Paralysis (HYPP), a disorder that causes episodes of weakness, fasciculation, and spasm, although not all heterozygotes are affected. The allele frequency of the variant in the halter subtype of Quarter Horses is 29.9% (15).
The allele frequency of the SCN4A variant exceeds the 5% threshold, although in this case it is clear that the variant shows incomplete penetrance.
GBE1: The GBE1 gene encodes the enzyme required for the synthesis of alpha 1-6 branches in glycogen. The GBE1 variant (GBE1-Y34X, a nonsense allele) is associated with Glycogen Branching Enzyme deficiency (GBED). GBED is a severe disease causing many foals to be aborted or stillborn; if foals survive they are typically euthanized by four months of age. Two apparently homozygous horses that were mature adults were identified in this study (1). The region of the variant had rather low coverage (4x – 6x) of sequencing reads. Resequencing identified one of the horses as heterozygous for the recessive variant rather than homozygous; quality DNA was not available for the other horse. The allele frequency of the GBE1 variant is 13% in Western pleasure horses, a subtype of Quarter Horses (15).
The allele frequency of the GBE1 variant, a recessive lethal with complete penetrance, exceeds the 5% threshold.
MUTYH: The MUTYH gene encodes an enzyme required for the repair of oxidative damage to DNA. Cerebellar abiotrophy is a neurodegenerative disease. Horses homozygous for the variant show a range of symptoms, with some horses apparently unaffected. Symptoms typically manifest in foals. The allele frequency of the MUTYH variant associated with cerebellar abiotrophy in Arabians (a mutation affecting the promoter) was 10% in this study (1).
The allele frequency of the MUTYH variant, which has incomplete penetrance, exceeds the 5% threshold.
PPIB: The PPIB gene encodes an enzyme that catalyzes the cis-trans isomerization of proline peptide bonds, potentially assisting in protein folding. A PPIB variant (PPIB-G39R) is associated with hereditary equine regional dermal asthenia (HERDA). HERDA is a severe disease affecting most horses by two years of age, when they are typically euthanized due to skin lesions. This study reports an allele frequency of 3% in Quarter Horses for this variant (1). The allele frequency of the PPIB variant is 14% in cutting horses, a subtype of Quarter Horses (15).
The allele frequency of the PPIB variant exceeds the 5% threshold.
GYS1: The GYS1 gene encodes glycogen synthase, the enzyme responsible for the synthesis of alpha 1-4 branches in glycogen. A variant allele, GYS1-R309H, is associated with Polysaccharide Storage Myopathy type 1 (16). While all horses carrying the variant exhibit constitutively activated glycogen synthase (17), many horses display no obvious symptoms. The allele frequency of the GYS1-R309H variant in this study is 10% in Belgians (1). The highest allele frequency reported is 14.2% in cutting horses, a subtype of Quarter Horses (15).
The allele frequency of the GYS1 variant exceeds the 5% threshold, although McCoy et al. (18) proposed that the GYS1-R309H variant was under positive selection for much of the history of domestication.
PLOD1: The PLOD1 gene encodes a lysyl oxidase required for the posttranslational modification of collagen. A variant allele is associated with Ehler-Danlos Syndrome, Type VI, previously known as Fragile Foal Syndrome. This is a disease affecting collagen quality. Many homozygotes die in utero, while for others this is a perinatal lethal. The allele frequency of the variant is 5.7% in the Warmbloods in this study (1). Another study reported a carrier frequency as high as 15% in some Warmblood subtypes; this corresponds to an allele frequency as high as 7.5% (9).
The allele frequency of the PLOD1 allele, a recessive lethal with complete penetrance, exceeds the 5% threshold.
MYOT: The MYOT gene encodes a Z disc protein that is part of a complex of actin-binding proteins. The P2 allele is a missense allele that changes a serine residue in the serine-rich region to a proline. The highest allele frequency of the P2 variant of MYOT in this study is 40.7% in Standardbreds (1). Supplementary Table 6 does not identify MYOT loss-of-function alleles present at an allele frequency greater than 5%.
FLNC: The FLNC gene encodes filamin C, a muscle-specific filamin that is part of a complex of actin-binding proteins at the Z disc. The P3 variant is a pair of missense alleles affecting the Ig-like repeats. The highest allele frequency of the FLNC variants is 8.6% in Thoroughbreds (1). Supplementary Table 6 does not identify FLNC loss-of-function alleles present at an allele frequency greater than 5%.
MYOZ3: The MYOZ3 gene encodes a Z disc protein that is part of a complex of actin-binding proteins. The P4 allele is a serine-to-leucine missense allele of a highly conserved position. The highest frequency of the P4 variant of MYOZ3 is 25% in Franches Montagnes; the next highest frequency is in Morgans at 9.1% (1). Supplementary Table 6 does not identify MYOZ3 loss-of-function alleles present at an allele frequency greater than 5%.
PYROXD1: The PYROXD1 gene encodes an oxidoreductase involved in the oxidative stress response. The P8 variant is a missense allele affecting a highly conserved position. The highest frequency of the P8 variant of PYROXD1 is 14.5% in Arabians (1). Supplementary Table 6 does not identify PYROXD1 loss-of-function alleles present at an allele frequency greater than 5%.
COL6A3: The COL6A3 gene encodes a collagen that is a component of the extracellular matrix in muscle. The K1 allele of COL6A3 is a glycine substitution in the triple helical region that is expected to be mildly damaging based on the analysis of human COL6A3 glycine substitutions. The highest allele frequency of the K1 variant of COL6A3 in this study is 8.5% in Standardbreds (1). Supplementary Table 6 does not identify COL6A3 loss-of-function alleles present at an allele frequency greater than 5%.
Table 6. Highest allele frequency of disease-associated variants
| Gene | Disease Association | Highest Frequency | Reference | Conclusion |
| PKHD1 | Congenital Liver Fibrosis | 89.5% | 1 | Eliminated |
| TRIM1-RPP2 | Juvenile Idiopathic Epilepsy | >50.0% | 1 | Eliminated |
| MYOT | Exercise intolerance | 40.7% | 1 | Proposed |
| SCN4A | Hyperkalemic Periodic Paralysis | 29.9% | 15 | Causative |
| MYOZ3 | Exercise intolerance | 25.0% | 1 | Proposed |
| PYROXD1 | Exercise intolerance | 14.5% | 1 | Proposed |
| PPIB | Hereditary Equine Regional Dermal Asthenia | 14.2% | 15 | Causative |
| GBE1 | Glycogen Branching Enzyme Deficiency | 13.0% | 15 | Causative |
| MUTYH | Cerebellar Abiotrophy | 10.0% | 1 | Causative |
| GYS1 | Polysaccharide Storage Myopathy | 10.0% | 1 | Causative |
| FLNC | Exercise intolerance | 8.6% | 1 | Proposed |
| COL6A3 | COL6-associated Myopathy | 8.5% | 1 | Proposed |
| PLOD1 | Fragile Foal Syndrome | 7.5% | 19 | Causative |
Evaluation of missense alleles
It is clear that some damaging variant alleles are present at allele frequencies in excess of 5% in some horse breeds or subtypes. The allele frequency of a variant is only one method of evaluating whether it is damaging, and while it is useful in the bulk analysis of genomes presented here (1), individual missense alleles can be evaluated by additional means.
One method for the evaluation of a missense allele is the comparison of the substituted amino acid for the amino acid in the reference allele. Amino acids can be grouped into categories based on their chemistry. For example, the substitution of any of the three branched-chain amino acids (leucine, isoleucine, and valine) for one of the others is unlikely to be damaging to protein structure or function, while the replacement of an amino acid by a chemically dissimilar one (e.g. leucine for serine, a hydrophobic amino acid for a charged amino acid) is more likely to be damaging.
A more powerful method of evaluating a missense allele is evolutionary conservation. In this approach, the reference and variant protein sequences are aligned to the sequence of the orthologous proteins from a wide range of species. If the substitution has gone to fixation in a species, it is not likely to be damaging. This is similar to the approach used in the current study to eliminate the PKHD1 and TRIM1-RPP2 variants as damaging, as they are present at high frequency in some breeds or subtypes (1).
This study does not systematically evaluate missense alleles present in horse genomes (1); the authors mention in passing the allele frequencies of the MYOT, MYOZ3, PYROXD1, FLNC, and COL6A3 variants. They have seen these variants evaluated using evolutionary conservation across mammals, birds, reptiles, amphibians, and fish, although they do not comment on this approach in their paper.
What is a Mendelian phenotype?
A Mendelian phenotype (named after Gregor Mendel) is one that results from mutation of a single gene. It is fully penetrant and is not influenced by variants of other genes or by environmental conditions. One example is albinism, seen in a wide range of species, which results from mutations in the gene encoding tyrosinase (TYR). Albinos show a complete absence of melanin; this phenotype is not affected by other genetic variants or by the environment.
A relatively small number of genetic variants are associated with phenotypes in horse. The easiest variants to discover are those that are Mendelian. In the examples summarized in Table 6, only the MUTYH and PLOD1 variants, responsible for Cerebellar Abiotrophy and Fragile Foal Syndrome, respectively, could be considered Mendelian. The SCN4A, PPIB, and GYS1 variants show incomplete penetrance.
The authors conclude that the missense alleles of MYOT, MYOZ3, PYROXD1, FLNC, and COL6A3 do not cause Mendelian disease. We agree with this conclusion. There are many examples in human genetics of phenotypes that are clinically indistinguishable caused by mutations in a number of different genes. None of these variants can be considered Mendelian. The analysis of genetic variation in horse, advanced greatly by the present study (1), will eventually reveal many components of the complex inheritance of the predisposition to muscle disease.
Conclusions
This study (1) is a landmark in the analysis of genetic variation in horse. We commend the researchers for the scale and thoroughness of this work. They have provided a wealth of data for other researchers to build upon. The analysis of genes affected by the 100 most frequent loss-of-function variants shown here is one small example of this sort of work, which points the way to improved annotation of the horse genome and the evaluation of transcript models. Will horses be recognized as a species that can provide insight into human genetic variation, particularly that affecting athletic performance? We think that this work is an important step in that direction.
References
1. Durward-Akhurst , SA, et al. (2024) Predicted genetic burden and frequency of phenotype-associated variants in the horse. Sci Rep. 14(1):8396. PMID: 38600096
2. Bosse, M, et al. (2018) Deleterious alleles in the context of domestication, inbreeding, and selection. Evol Appl 12(1):6-17. PMID: 30622631
3. Bertorelle, G, et al. (2022) Genetic load: genomic estimates and applications in non-model animals. Nat Rev Genet. 23(8):492-503. PMID: 35136196
4. Dussex, N, et al. (2023) Purging and accumulation of genetic load in conservation. Trends Ecol Evol. 38(10):961-969. PMID: 37344276
5. Robinson, J, et al. (2023) Deleterious variation in natural populations and implications for conservation genetics. Annu Rev Anim Biosci. 11:93-114. PMID: 36332644
6. Kryukov, G, et al. (2007) Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. Am J Hum Genet. 80(4):727-739. PMID: 17357078
7. MacArthur, DG, et al. (2012) A systematic survey of loss-of-function variants in human protein-coding genes. Science. 335(6070):823-828. PMID: 22344438
8. Derks et al. (2018) Balancing selection on a recessive lethal deletion with pleiotropic effects on two neighboring genes in the porcine genome. PLoS Genet. 14(9):e1007661. PMID: 30231021
9. Derks et al. (2018) A survey of functional genomic variation in domesticated chickens. Genet Sel Evol 50: 17. PMID: 29661130
10. Drögemüller M et al. (2014) Congenital Hepatic Fibrosis in the Franches-Montagnes horse is associated with the Polycystic Kidney and Hepatic Disease 1 (PKHD1) Gene. PLoS One 9(10):e110125. PMID: 25295861
11. Molín J et al. (2018) Congenital Hepatic Fibrosis in a Purebred Spanish Horse Foal: Pathology and Genetic Studies on PKHD1 Gene Mutations. Vet Pathol. 55(3):457-461. PMID: 29402207
12. Polani S et al. (2022) Sequence variant in the TRIM39-RPP21 gene readthrough is shared across a cohort of Arabian foals diagnosed with juvenile idiopathic epilepsy. J Genet Mutat Disord. 1(1):103. PMID: 35465405
13. Rivas VN et al. (2019) TRIM39-RPP21 variants (∆19InsCCC) are not associated with juvenile idiopathic epilepsy in Egyptian Arabian horses. Genes (Basel). 10(10):816. PMID: 31623255
14. Aleman et al. (2018) Investigation of known genetic mutations of Arabian horses in Egyptian Arabian foals with juvenile idiopathic epilepsy. J Vet Intern Med. 32(1):465-468. PMID: 29171123
15. Tyron, RC et al. (2009) Evaluation of allele frequencies of inherited disease genes in subgroups of American Quarter Horses. J Am Vet Med Assoc. 234(1):120-5. PMID: 19119976
16. McCue ME et al. (2008) Glycogen synthase (GYS1) mutation causes a novel skeletal muscle glycogenosis. Genomics. 91(5):458-66. PMID: 18358695
17. Maile CA et al. (2017) A highly prevalent equine glycogen storage disease is explained by constitutive activation of a mutant glycogen synthase. Biochim Biophys Acta Gen Subj. 861(1 Pt A):3388-3398. PMID: 27592162
18. McCoy et al. (2014) Evidence of positive selection for a glycogen synthase (GYS1) mutation in domestic horse populations. J Hered. 105(2):163-72. PMID: 24215078
19. Wobbe M et al. (2022) Quantifying the effect of Warmblood Fragile Foal Syndrome on foaling rates in the German riding horse population. PLOS One 17(7):e0267975. PMID: 35901076