
Blog
The K1 Genetic Variant Affects COL6A3, a Gene Encoding a Collagen
13 Nov 2020

Contents
- Introduction
- The collagen triple helix
- Figure 1. Primary structure of the human COL6A3 protein
- Figure 2. The collagen triple helix
- Bethlem and Ullrich myopathy
- Severity of the disease is correlated with the position of the amino acid substitution
- Table 1. Pathogenic and likely pathogenic missense alleles of human COL6A3
- Figure 3. Alignment of the triple helical regions of equine and human COL6A3 protein sequences
- Effects of the K1 allele of COL6A3 in horses
- Table 2. Genotype of the Paint Horse in which the K1 allele of COL6A3 was identified
- References
Introduction
The K1 genetic variant that has been part of EquiSeq’s Myopathy Panel since October 2019 is a missense allele of COL6A3, a gene encoding a collagen [1]. Collagens are a family of proteins that are the main structural components of the connective tissue. Collagens also guide bone formation and play an important role in many other tissues. They are the most abundant proteins in the body of mammals.
There are thirty types of collagen, made up of collagen proteins encoded by 44 different genes. In humans, mutations in different collagen genes are responsible for a wide variety of inherited disorders. The phenotype produced by mutations in different collagens gives us an insight into the role of each of these collagens.
The COL6A3 gene encodes one of the type VI collagens that are components of the endomysium, a layer of connective tissue that covers individual muscle fibers. Mutations in human COL6A1, COL6A2, and COL6A3 are associated with Bethlem myopathy and Ullrich congenital muscular dystrophy [1-11].
The K1 allele of equine COL6A3 is defined by its coordinates in the current genome assembly for horse, chr6:23,416,882 C/G in EquCab3.0 [12]. For purposes of this blog post, we will use the protein model XP_023498413.1, which would make this allele G2178A, the substitution of an alanine (A) for a glycine (G) in amino acid position 2178.
The collagen triple helix
Proteins are polymers of twenty different amino acids that are defined by both their primary structure (the sequence of amino acids that make up the protein) as well as by their secondary structure (the way that the protein folds in three dimensions).
Figure 1 shows the primary structure of the COL6A3 protein.
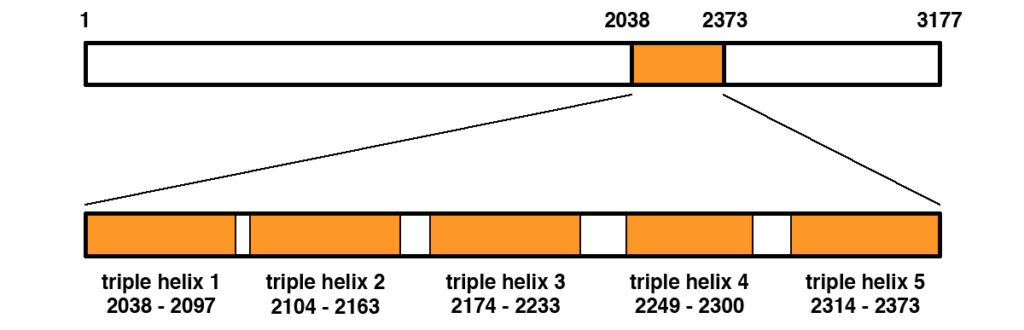
Figure 1. Primary structure of the human COL6A3 protein (NP_004360.2). The protein is 3177 amino acids in length. A middle portion spanning amino acids 2038 to 2373 contains the triple helical region that becomes part of the extracellular matrix once exported from the cell. The triple helical region contains five regions of triple helix as shown, separated by short interdomain regions.
The collagen triple helix is a secondary structure of different types of collagen. It consists of three different collagen molecules wrapped around each other in a helical fashion as shown in Figure 2.
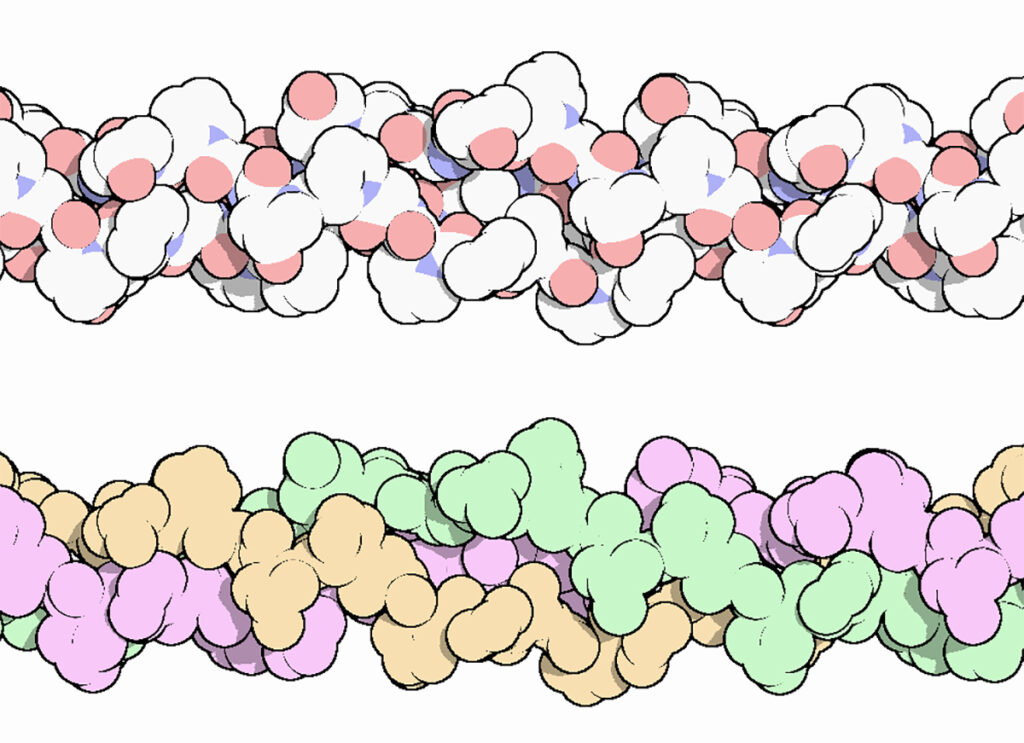
Figure 2. The collagen triple helix, colored by atom, with oxygen in red and nitrogen in blue (top), and colored to highlight the three chains (bottom). Three different protein molecules are wrapped around each other in a helical fashion.
The portion of the collagen molecule that participates in triple helix formation is made up of a repetitious sequence of amino acids with the general sequence Gly-X-Y. Every third residue is a glycine. This is critical to the structure, because glycine, with an R group consisting of a single hydrogen, is the only amino acid that can have contact with the center of the triple helix due to spatial constraints. The collagen triple helix is also rich in proline and hydroxyproline residues at the X and Y positions. The large number of proline and hydroxyproline residues prevent the formation of regular alpha helix and beta sheets, which are common secondary structures found in other proteins.
Bethlem and Ullrich myopathy
Missense alleles of COL6A1, COL6A2, and COL6A3 are responsible for Bethlem myopathy and Ullrich congenital muscular dystrophy [3 – 11]. Bethlem myopathy was originally described as a mild disease associated with dominant alleles of these three genes, while Ullrich congenital muscular dystrophy was originally described as a more severe disease associated with recessive alleles. Some patients with the mild form of the disease have since been found to be compound heterozygotes for two different alleles of the same gene, while dominant alleles have been found to be associated with the severe form. Ullrich congenital muscular dystrophy has been reclassified as a form of limb-girdle muscular dystrophy (LGMDR22) [13].
Human patients with defects in COL6A1, COL6A2, or COL6A3 associated with Bethlem or Ullrich myopathy have an array of symptoms including muscle weakness, muscle contractures, muscle atrophy, hyperextensibility in distal joints, rigid spine, abnormal scar tissue, and difficulties in ambulation [7, 11]. Patients who die of the disease often die as a result of respiratory insufficiency resulting from weakness in the diaphragm, which is skeletal muscle.
Severity of the disease is correlated with the position of the amino acid substitution
The severity of alleles of COL6A3 is correlated with their position in the collagen triple helix. Alleles associated with disease that affect the triple helix region are missense alleles that replace one of the required glycine residues of the Gly-X-Y repeat with a different amino acid. Substitutions of glycine residues near the amino-terminal portion of the triple helix have a more severe effect than substitutions near the carboxy-terminal portion of the triple helix, suggesting that assembly of the triple helix is initiated in the amino-terminal region. Pathogenic human COL6A3 alleles are summarized in Table 1.
Table 1. Pathogenic and likely pathogenic missense alleles of human COL6A3.
| Allelea | Pathogenicityb | Domainc | Cited ind |
|---|---|---|---|
| G2053V | Pathogenic | Triple helix | ClinVar |
| G2053C | Pathogenic | Triple helix | ClinVar, EGL |
| G2056E | Likely pathogenic | Triple helix | EGL |
| G2059C | Pathogenic | Triple helix | ClinVar, EGL |
| G2065R | Pathogenic | Triple helix | ClinVar, EGL |
| G2065S | Likely pathogenic | Triple helix | ClinVar, EGL |
| G2071D | Likely pathogenic | Triple helix | ClinVar, EGL |
| G2074S | Pathogenic | Triple helix | ClinVar, EGL |
| G2077V | Pathogenic | Triple helix | ClinVar |
| G2077D | Pathogenic | Triple helix | ClinVar, EGL |
| G2080D | Pathogenic | Triple helix | ClinVar, EGL |
| G2080R | Pathogenic | Triple helix | ClinVar |
| G2080S | Likely pathogenic | Triple helix | ClinVar, EGL |
| G2083V | Likely pathogenic | Triple helix | ClinVar |
| G2098V | Pathogenic | Interdomain | ClinVar, EGL |
| K2103N | Likely pathogenic | Interdomain | ClinVar |
| K2103R | Likely pathogenic | Interdomain | ClinVar |
| G2140R | Likely pathogenic | Triple helix | ClinVar |
| G2267S | Likely pathogenic | Triple helix | EGL |
| G2285R | Likely pathogenic | Triple helix | ClinVar |
| G2297A | Likely pathogenic | Triple helix | EGL |
aMissense alleles are described by the amino acid in the reference sequence, the amino acid position in the human COL6A3 protein (NP_004360.2), and the amino acid in the missense allele. For example, G2053V is the substitution of a valine (V) for a glycine (G) at amino acid position 2053.
bPathogenicity is given for missense alleles judged to be pathogenic or likely pathogenic in the two databases listed based on clinical evidence.
cAll missense alleles identified as pathogenic or likely pathogenic between positions 2038 and 2373 of human COL6A3 protein (NP_004360.2), as shown in Figure 1, are listed. Most of these alleles affect segments of triple helix, but three alleles fall into an interdomain region.
dDatabases used are ClinVar and EGL Genetics.
Figure 3 shows the alignment of the triple helical region of the equine and human COL6A3 protein sequences and the position and amino acid substitutions observed in pathogenic human COL6A3 alleles and in the K1 allele of equine COL6A3.
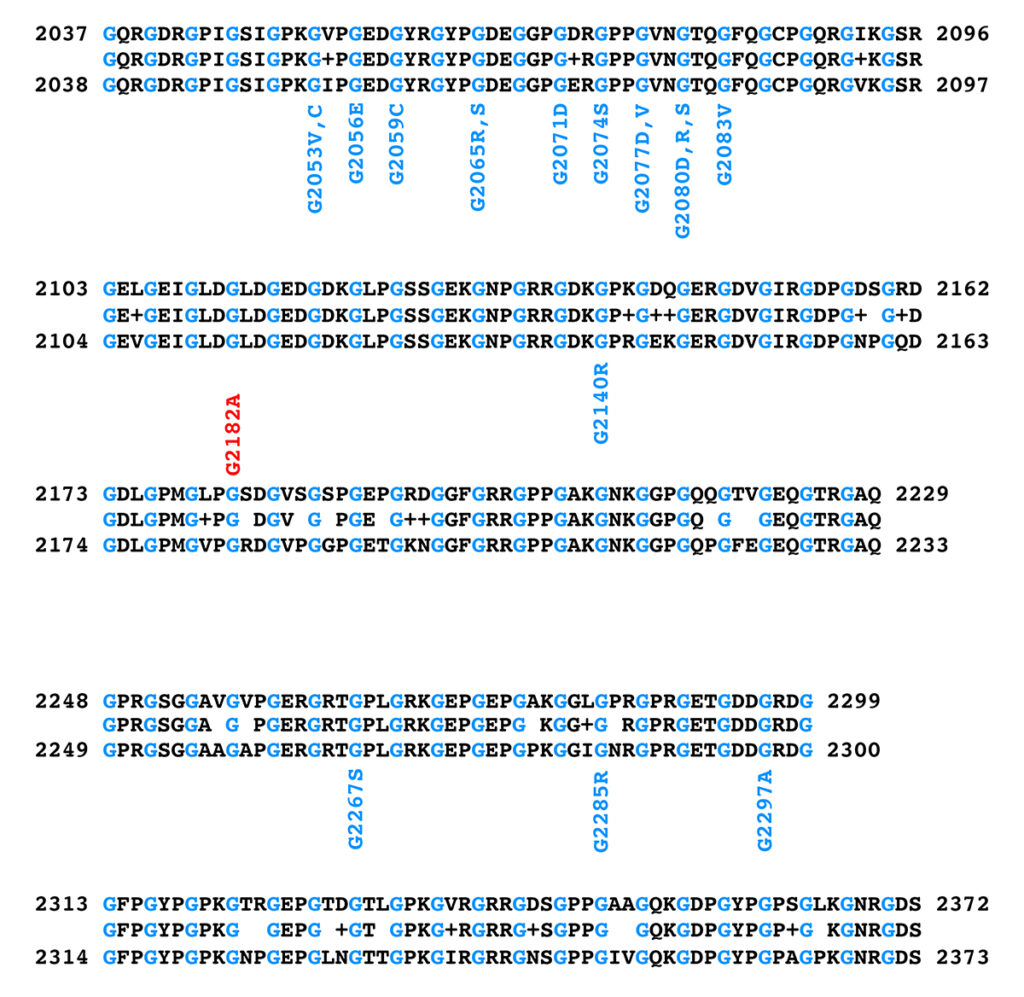
Figure 3. Alignment of the triple helical regions of equine (top) and human (bottom) COL6A3 protein sequences. The equine sequence is from XP_023498413.1, while the human sequence is from NP_004360.2. The sequence between the equine and human sequences in the alignment shows the amino acid if the equine and human sequences at that position are identical, a “+” if the amino acids at that position differ by a conservative substitution, and a blank space if the amino acids at that position differ by a nonconservative substitution. Glycine residues are shown in blue in the sequences to highlight the Gly-X-Y repeat structure of the triple helical region. Note that the glycine residues are all identical between the equine and human sequences. Positions and amino acid substitutions of pathogenic human missense alleles are shown in blue below the human sequence; the equine K1 allele is shown in red above the equine sequence.
The work on human alleles of COL6A3 suggests that the K1 allele of equine COL6A3 should have a moderate semidominant effect and a more severe defect in homozygotes. There is no effective treatment for human patients with Bethlem myopathy or Ullrich congenital muscular dystrophy.
Effects of the K1 allele of COL6A3 in horses
The K1 allele of COL6A3 was originally identified by whole genome sequencing of a Paint Horse with symptoms of exercise intolerance and a diagnosis of Polysaccharide Storage Myopathy type 2 by muscle biopsy at four years of age. Symptoms first appeared at one year of age. This horse is heterozygous for K1 (n/K1). The horse is homozygous for the wild-type alleles of all five genetic variants in the Quarter Horse “five panel,” and also for the wild-type alleles of other genes included in EquiSeq’s Myopathy Panel, as shown in Table 2.
Table 2. Genotype of the Paint Horse in which the K1 allele of COL6A3 was identified.
| Allelea | Nicknameb | Disease statec | Genotyped |
|---|---|---|---|
| SCN4A-F1416L | H | HYPP | n/n |
| PPIB-G39R | Hrd | HERDA | N/N |
| GBE-T34X | Gb | GBED | N/N |
| RYR1-R2454G | MH | MH | n/n |
| GYS1-R309H | P1 | PSSM1 | n/n |
| MYOT-S232P | P2 | PSSM2 | n/n |
| FLNC-E753K | P3a | PSSM2/MFM | n/n |
| FLNC-A1207T | P3b | PSSM2/MFM | n/n |
| MYOZ3-S42L | P4 | PSSM2/MFM | n/n |
| PYROXD1-D492H | P8 | PSSM2/MFM | n/n |
| COL6A3-G2178A | K1 | PSSM2 | n/K1 |
aAlleles are described using the gene symbol and the amino acid substitution based on the commonly accepted protein model. All are missense alleles except for GBE-T34X, which is a premature termination allele.
bNicknames are substitute allele symbols in common usage among horse owners.
cDisease states are: Hyperkalemic Periodic Paralysis (HYPP), Hereditary Equine Regional Dermal Asthenia (HERDA), Glycogen Branching Enzyme Deficiency (GBED), Malignant Hyperthermia (MH), Polysaccharide Storage Myopathy, type 1 (PSSM1), Polysaccharide Storage Myopathy, type 2 (PSSM2), Myofibrillar Myopathy (MFM).
dN or n denotes the wild-type allele.
This n/K1 horse exhibits fasciculations of the triceps musculature in the front limbs after moderate exercise. After moderate exercise the horse becomes resistant to moving forward and exhibits a gait abnormality termed “rope walking,” where the striding leg is twisted around in front of the supporting leg, as if the horse is walking a tightrope. The horse does not show this gait abnormality when rested.
A second muscle biopsy was performed at eight years of age. Frozen sections were immunostained with antibodies to human COL6A3 and the basement membrane protein perlecan. There was no sign of endomysial fibrosis, a finding similar to that seen in human patients with moderate Bethlem myopathy [8, 9, 14, 15]. Scar tissue is normal with no evidence of keloid formation, and there is no overt sign of hyperextension or contractures of the distal limbs.
A survey of different breeds from a sample unselected for symptoms of exercise intolerance shows that the K1 allele of COL6A3 is absent from Cleveland Bay (23 horses tested), Mangalarga Marchadore (24), Turkoman (24), Selle Francois (18), Suffolk Punch (24), Tennessee Walker (24), Shetland Pony (24), and Shire (24). The K1 allele of COL6A3 is present in Arabians (3 heterozygotes in 24 horses for an allele frequency of 3/48), Fell Pony (2/46), Mangalarga (1/50), Campolina (1/48), Caspian (3/48), Kurd (3/48), Akhal Teke (2/48), Highland Pony (5/48), and Exmoor Pony (6/52). The same sample set contained two breeds in which K1/K1 homozygotes were observed; one K1/K1 and 7 n/K1 horses were seen in a sample of 24 Haflingers (allele frequency 9/48) and one K1/K1 and 5 n/K1 horses were observed among 24 Standardbreds (allele frequency 7/48). While these samples are relatively small, the results show that the K1 allele is widely distributed among unrelated breeds.
The unselected samples described above were from archived DNA from a breed survey. No homozygous horses (K1/K1) have been directly observed by researchers at EquiSeq.
Samples received at EquiSeq that cannot be considered to be unselected show that the K1 allele of COL6A3 is rare among stock breeds (present in only two n/K1 horses in 242 Quarter Horses, Paints, and Appaloosas despite having been discovered in a Paint horse) and absent in a sample of 144 Thoroughbreds. It is present in Arabians (allele frequency 24/488), Haflingers (5/40), Standardbreds (2/44), and Miniatures (2/18). We are in the process of surveying additional horses, especially Haflingers and Standardbreds, in order to attempt to find a homozygote (K1/K1).
References
[1] Chu M.-L et al. (1990). Mosaic structure of globular domains in the human type VI collagen alpha-3 chain: similarity to von Willebrand factor, fibronectin, actin, salivary proteins and apotinin type protease inhibitors. EMBO J. 9: 385-393. PMID: 1689238
[2] Chu M.-L et al. (1989). Sequence analysis of alpha 1(VI) and alpha 2(VI) chains of human type VI collagen reveals internal triplication of globular domains similar to the A domains of von Willebrand factor and two alpha 2(VI) chain variants that differ in the carboxy terminus. EMBO J. 8(7): 1939-1346. PMID: 2551668
[3] Pan T-C et al. (1998). Missense mutation in a von Willebrand factor type A domain of the alpha 3(VI) collagen gene (COL6A3) in a family with Bethlem myopathy. Hum Mol Genet. 7(5): 807-812. PMID: 9536084
[4] Pepe G et al. (1999). A novel de novo mutation in the triple helix of the COL6A3 gene in a two-generation Italian family affected by Bethlem myopathy. A diagnostic approach in the mutations’ screening of type VI collagen. Neuromuscul Disord. 9(4): 264-271. PMID: 10399756
[5] Jöbsis GJ et al. (1999). Bethlem myopathy: a slowly progressive congenital muscular dystrophy with contractures. Brain. 122(4): 649-655. PMID: 10219778
[6] Baker NL et al. (2004). Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum Mol Genet. 14(2): 279-293. PMID: 15563506
[7] Lampe AK, Bushby KMD (2005). Collagen VI related muscle disorders. J Med Genet. 42(9): 673-685. PMID: 16141002
[8] Pace RA et al. (2008). Collagen VI glycine mutations: Perturbed assembly and a spectrum of clinical severity. Ann Neurol. 64(3): 294-303. PMID: 18825676
[9] Briñas L et al. (2010). Early onset collagen VI myopathies: Genetic and clinical correlations. Ann Neurol. 68(4): 511-520. PMID: 20976770
[10] Butterfield RJ et al. (2013). Position of glycine substitutions in the triple helix of COL6A1, COL6A2, and COL6A3 is correlated with severity and mode of inheritance in collagen VI myopathies. Hum Mutat. 34(11): 1558–1567. PMID: 24038877
[11] Kirschner J (2013). Congenital muscular dystrophies. Handb. Clin. Neurol. 113: 1377-1385. PMID: 23622361
[12] Kalbfleisch TS et al. (2018). Improved reference genome for the domestic horse increases assembly contiguity and composition. Commun Biol. 1: 197. PMID: 30456315
[13] Straub V et al. (2018). 229th ENMC international workshop: limb girdle muscular dystrophies–nomenclature and reformed classification, Naarden, the Netherlands, 17-19 March 2017. Neuromusc. Disord. 28: 702-710. PMID: 30055862
[14] Lamandé SR et al. (2002). Kinked Collagen VI Tetramers and Reduced Microfibril Formation as a Result of Bethlem Myopathy and Introduced Triple Helical Glycine Mutations. J Biol Chem. 277(3): 1949-1956. PMID: 11707460
[15] Hicks, D et al. (2008). A refined diagnostic algorithm for Bethlem myopathy. Neurology. 70(14): 1192-1199. PMID: 18378883
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
Carnegie Mellon University Profiles EquiSeq Founder
April 20, 2026 – Pittsburgh, Pennsylvania The alumni magazine for Carnegie Mellon published…
22 Apr 2026
EquiSeq hosts webinar on Recurrent Exertional Rhabdomyolysis (RER)
ALBUQUERQUE, NEW MEXICO – April 4, 2026 EquiSeq hosted a webinar on Recurrent…
4 Apr 2026
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
