
Blog
The Px Allele of CACNA2D3 May Affect Splicing of a Component of the Voltage-Gated Calcium Channel
8 Feb 2020

A Genome Wide Association Study (GWAS) of Recurrent Exertional Rhabdomyolysis (RER) identified a region on chromosome 16 as the only part of the genome associated with RER [1]. One of the populations in the study showed an association between RER and five Single Nucleotide Polymorphisms (SNPs) that define a 71.6 kb interval within the 822 kb CACNA2D3 gene. The CACNA2D3 gene encodes a regulatory subunit of the voltage-gated calcium channel called the dihydropyridine receptor (DHPR) that functions in excitation-contraction coupling, the process through which signals from motor neurons initiate muscle contraction [2].
The alpha(2)delta subunit encoded by CACNA2D3 sets the abundance of voltage-gated calcium channels by acting as a molecular chaperone. The alpha(2)delta subunit is bound to the plasma membrane through a glycosylphosphatidylinositol (GPI) anchor [3,4]. The level of expression of alpha(2)delta subunits sets the abundance of voltage-gated calcium channels in both neuronal and transfected nonneuronal cells [5,6].
The dihydropyridine receptor, of which the CACNA2D3 protein is a part, is known to physically interact with the ryanodine receptor encoded by the RYR1 gene. Malignant Hyperthermia (MH) in horses is caused by mutations in the RYR1 gene [7]. The RYR1-R2454G allele that causes MH in horses acts as an enhancer of the GYS1-R309H allele of glycogen synthase that is associated with Polysaccharide Storage Myopathy, Type 1 (PSSM1) [8]. It is therefore plausible that mutations in CACNA2D3 might both cause exercise intolerance on their own, as well as potentially acting as enhancers of other genetic variants that cause exercise intolerance.
We surveyed the CACNA2D3 gene in the genomes of fourteen horses for which we obtained whole genome sequences. These horses include two Thoroughbreds with symptoms of exercise intolerance that test negative for GYS1-R309H (P1), MYOT-S232P (P2), FLNC-E753K + FLNC-A1207T (P3), and MYOZ3-S42L (P4). There are no nonsynonymous substitutions (missense alleles, nonsense alleles, stop-loss alleles, and start-loss alleles) or frameshifts in the entire coding region of CACNA2D3 in any of these horses. We found four synonymous substitutions, shown below.
| TABLE 1. Synonymous Substitutions in CACNA2D3 | ||||
|---|---|---|---|---|
| Namea | Exon | Coordinateb | Variantc | Codon Changed |
| A525A (Px) | 20 | chr16: 33,132,988 | C/T | GCG -> GCA |
| T179T | 7 | chr16: 33,407,225 | C/T | ACG -> ACA |
| K226K | 8 | chr16: 33,395,933 | C/T | AAG -> AAA |
| F672F | 25 | chr16: 33,118,160 | G/A | TTC -> TTT |
| aThe allele name shows the reference amino acid, the codon, and the new amino acid (always the same for a synonymous substitution). | ||||
| bCoordinates are given for EquCab2.0. Note that in EquCab3.0 [9], the Px allele is chr16: 34,635,494 T/C, that is, the allele that we have designated as Px is now the reference allele for this SNP. | ||||
| cThe reference allele is given first; the allele considered the variant is given after the slash. Note that in EquCab3.0 [9], the Px allele is chr16:34,635,494 T/C, that is, the allele that we have designated as Px is now the reference allele for this SNP. | ||||
| dBecause CACNA2D3 is on the reverse strand, the codon change shows the complement of the variant, which is given for the forward strand. | ||||
The inheritance of three of the synonymous substitutions is not correlated with the inheritance of variant alleles of the SNPs identified in the GWAS study [Fritz KL 2012]. The inheritance of a single synonymous substitution, CACNA2D3-A525A (16:33132988 C/T in EquCab 2.0), is absolutely correlated with the inheritance of variant alleles of three of the five SNPs from the GWAS study (SNPs 1-3 in Table 2), and somewhat correlated with the inheritance of variant alleles of the other two SNPs (SNPs 4 and 5 in Table 2). These five SNPs are shown in Table 2 below [1].
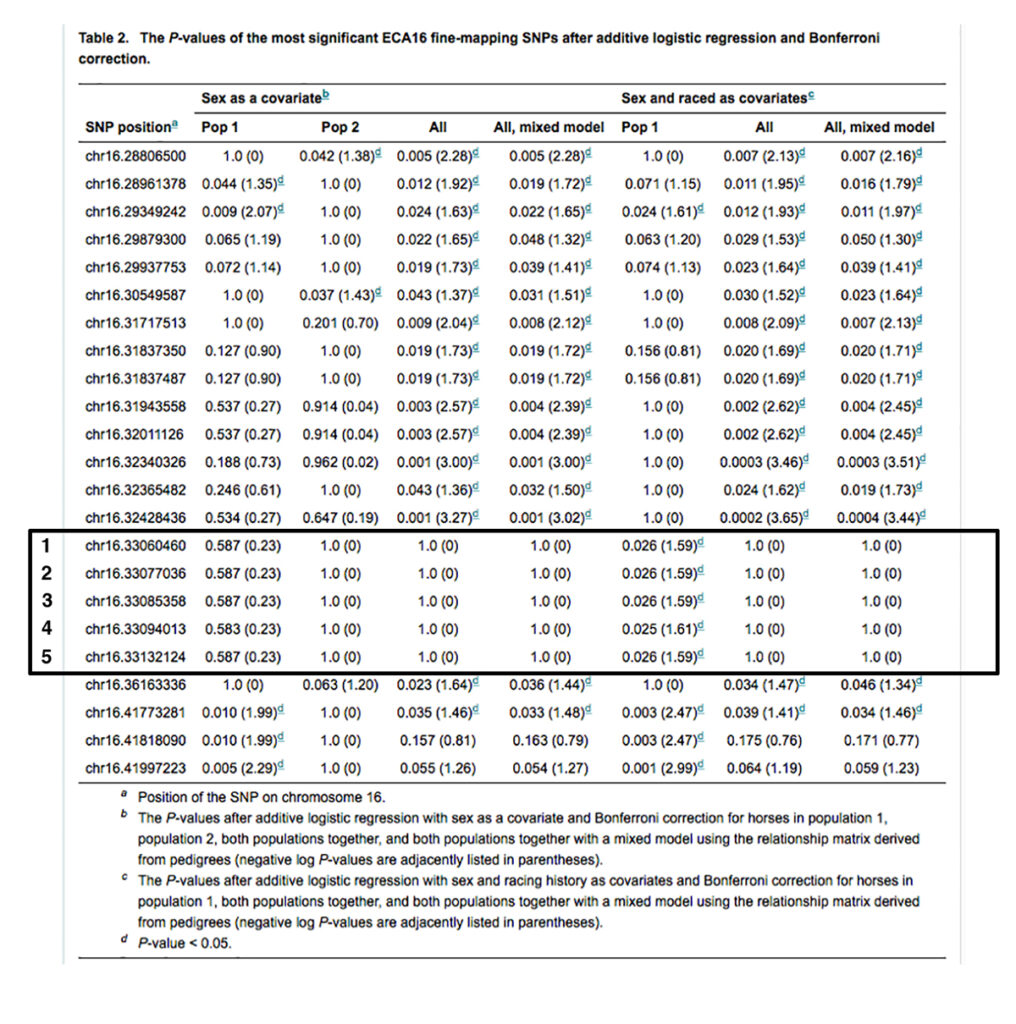
The CACNA2D3-A525A variant, termed Px, is heterozygous in the two exercise-intolerant Thoroughbreds free of the P1, P2, P3, and P4 variants; it is also heterozygous in an exercise-intolerant Appendix Quarter horse that is n/P1 n/P2 and another exercise-intolerant Quarter Horse that is n/P2.
Synonymous substitutions and exonic splicing enhancers
The protein-coding regions of genes specify which of the twenty primary amino acids occupy a specific position in a protein using a triplet code of three nucleotides (the genetic code). Because there are 64 possible three-nucleotide codons made up of A, T, C, and G, and only twenty primary amino acids, most amino acids can be specified by more than one codon. A mutation that changes a codon encoding a particular amino acid to a different codon encoding the same amino acid is called a synonymous substitution. Synonymous substitutions are generally considered selectively neutral, but there are exceptions.
The protein-coding region of most genes in mammals is interrupted by noncoding segments called introns. The primary transcript of a protein-coding gene is processed to remove introns, leaving only the coding regions (exons) in the mature mRNA. This means that in addition to the genetic code specifying the amino acids in the exons, there are also sequences in the primary transcript that signal the beginning and end of introns to be removed by splicing. These sequences are primarily at the beginning and ends of introns and are rather simple. For genes with a primary transcript that is large relative to the coding segment, there will be many possible sequences resembling legitimate splice sites that, if used, would give rise to an altered mature mRNA encoding a different form of the protein.
In some cases, synonymous substitutions are not selectively neutral, but rather have phenotypic effects [10, 11, 12, 13, 14, 15, 16, 17]. There are many more potential introns than are actually used in the splicing of the primary transcript of any particular gene, so it follows that there must be other signals that identify actual introns from the many potential ones. One such class of elements is Exonic Splicing Enhancers or ESEs. These are signals within the exons that recruit splicing factors to carry out the splicing reaction [13, 17]. Exonic Splicing Enhancers serve as the binding sites for SR proteins, part of the 100 or so proteins that make up the spliceosome.
Fairbrother et al. (2002) carried out a statistical analysis of 4817 human genes containing 31,463 introns and 28,933 internal exons [11]. They limited their analysis to the identification of hexamers, which is comparable in size to the binding sites of many known RNA binding factors. Analysis of each of the 4096 oligonucleotide hexamers identified 103 different hexamers as candidate 5 ́ ESEs and 198 different hexamers as candidate 3 ́ ESEs. There are 63 of the 103 5 ́ ESEs present in the set of predicted 3 ́ ESEs. A total of 238 of the 4096 possible hexamers are predicted to display either 5 ́ ESE or 3 ́ ESE activity, about 6% of the total. The authors have provided a public web-based platform for the identification of ESEs in submitted exon sequences.
We have evaluated the four synonymous substitutions seen in exons 7, 8, 20, and 25 of CACNA2D3 for the possibility that they might disrupt Exonic Splicing Enhancers. The figure below shows the sequence of these four exons and parts of the flanking introns. The locations of each of the four SNPs identified as synonymous substitutions are indicated by underlining. The figure shows the protein sequences fully encoded by the exons as well as all ESEs as detected by the public web-based platform [11].

As shown in the figure above, exon 20, containing the Px variant, is unusual in that there is only a single hexamer identifiable as an ESE located six bases from the site of the Px base substitution near the 5´end of the exon. Note that the ESE located at the 3´end of exon 20 consists of a set of four overlapping hexamers, each of which is known to be an ESE. This suggests that the ESE near the 5´end of exon 20 is much less robust, and potentially more vulnerable to disruption by a base substitution, than is the robust ESE near the 3´end of exon 20.
As shown in the figure above, the synonymous substitution in exon 7 is located far from the potential ESEs at the 5´end and 3´end of exon 7. The synonymous substitution in exon 8 alters a base in a robust ESE (five overlapping hexamers) at the 3´end of exon 8. The synonymous substitution in exon 25 is located far from the potential ESEs at the 5´end and 3´end of exon 25.
It is possible that the Px variant is not causative, but rather, like the SNPs with which it is associated, it is associated with another causative variant through linkage disequilibrium. We have not exhaustively examined intron variants seen in affected horses. The equine genomes examined so far are free of variants in CACNA2D3 that alter the most highly conserved regions that define an intron (the GT dinucleotide at the 5´end and the AG dinucleotide at the 3´end). A search for intron variants would be warranted if Px/Px homozygotes are not more severely affected, or if the association between Px and exercise intolerance breaks down in a larger sample.
Recommendations
A large number of horses have been tested for the Px variant since our original finding. Additional horses have been analyzed by whole genome sequencing as well. We have found apparently asymptomatic horses that are n/Px and Px/Px. We have also analyzed horses with clinically confirmed RER that lack the Px variant. The GWAS study suggests that if the Px variant is a genetic component of RER, it requires an additional genetic factor or factors to cause a phenotype [1].
We have included the Px allele in EquiSeq’s Myopathy Panel as a way of surveying the incidence of this genetic variant in as many horses as possible. We have received reports of n/Px and Px/Px horses that are clear for the other variants in the current Myopathy Panel (P2, P3, P4, P8, and K1) that are symptomatic, typically described as excitable. We have also received reports of horses that are n/Px and Px/Px that are clear for other variants that are considered asymptomatic by their owners.
The original GWAS study of RER found that the SNPs in linkage disequilibrium with the Px allele are only significant in a subgroup of related horses, suggesting that there may be another genetic component [1]. The observations shared with us from horse owners suggest that the Px allele is not always associated with symptoms. The Px allele remains in EquiSeq’s Myopathy Panel as an investigational variant. We do not recommend that horse owners use Px test results to guide a breeding program in horses that appear to be free of symptoms.
Citations
[1] Fritz KL et al. (2012). “Genetic mapping of recurrent exertional rhabdomyolysis in a population of North American Thoroughbreds.” Anim Genet. 43(6):730-738. PMID: 22497487.
[2] Arias-Calderón M et al. (2016). “Characterization of a multiprotein complex involved in excitation-transcription coupling of skeletal muscle.” Skelet Muscle. 6:15. PMID: 27069569.
[3] Gao B et al. (2000). “Functional properties of a new voltage-dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2).” J Biol Chem. 275(16):12237-12242. PMID: 10766861.
[4] Davies A et al. (2010). “The alpha2delta subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function.” Proc Natl Acad Sci U S A. 107(4):1654-1659. PMID: 20080692.
[5] Cao YQ et al. (2004). “Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy.” Neuron. 43(3):387-400. PMID: 15294146.
[6] Hoppa MB et al. (2012). “α2δ expression sets presynaptic calcium channel abundance and release probability.” Nature. 486(7401):122-125. PMID: 22678293.
[7] Aleman M et al. (2004). “Association of a mutation in the ryanodine receptor 1 gene with equine malignant hyperthermia.” Muscle Nerve. 30(3):356-365. PMID: 15318347.
[8] McCue ME et al. (2009). “Polysaccharide storage myopathy phenotype in quarter horse-related breeds is modified by the presence of an RYR1 mutation.” Neuromuscul Disord. 19(1):37-43. PMID: 19056269.
[9] Kalbfleisch TS et al. (2018). “Improved reference genome for the domestic horse increases assembly contiguity and composition.” Commun Biol. 1:197. PMID: 30456315.
[10] Cartegni L et al. (2002). “Listening to silence and understanding nonsense: exonic mutations that affect splicing.” Nat Rev Genet. 3(4):285-298. PMID: 11967553.
[11] Fairbrother WG et al. (2002). “Predictive identification of exonic splicing enhancers in human genes.” Science. 297(5583):1007-1013. PMID: 12114529.
[12] Maquat LE et al. (2001). “The power of point mutations.” Nat Genet. 27(1):5-6. PMID: 11137984.
[13] Pagani F and Baralle FE (2004). “Genomic variants in exons and introns: identifying the splicing spoilers.” Nat Rev Genet. 5(5):389-396. PMID: 15168696.
[14] Pagani F et al. (2005). “Genomic variants in exons and introns: identifying the splicing spoilers.” Nat Rev Genet. 5(5):389-396. PMID: 15840711.
[15] Carlini DB and Genut JE (2006). “Synonymous SNPs provide evidence for selective constraint on human exonic splicing enhancers.” J Mol Evol. 62(1):89-98. PMID: 16320116.
[16] Goren A et al. (2006). “Comparative analysis identifies exonic splicing regulatory sequences–The complex definition of enhancers and silencers.” Mol Cell. 22(6):769-781. PMID: 16793546.
[17] Baralle D and Baralle M (2015). “Splicing in action: assessing disease causing sequence changes.” J Med Genet. 42:737-748. PMID: 16199547.
Share this post
From the blog
The latest industry news, interviews, technologies, and resources.
EquiSeq hosts webinar on Recurrent Exertional Rhabdomyolysis (RER)
ALBUQUERQUE, NEW MEXICO – April 4, 2026 EquiSeq hosted a webinar on Recurrent…
4 Apr 2026
EquiSeq Profiled on KRQE
ABQ startup hopes to end horse disease with genetic research and testing. View…
11 Mar 2026
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
