Our blog
Resources and insights
The latest industry news, interviews, technologies, and resources.
Blog categories
News
Jeremy Edwards speaks on intellectual property
Jeremy Edwards, Chair of Department of Chemistry and Chemical Biology at the University of New Mexico, is one of UNM’s most prolific inventors. Dr. Edwards is part of the EquiSeq science team and is an inventor on two of EquiSeq’s patents. In this interview, Dr. Edwards talks about the importance of protecting intellectual property.
3 Dec 2024
News
South Africa issues patent on EquiSeq DNA tests
ALBUQUERQUE, NEW MEXICO EquiSeq announced today that South Africa has issued a patent on EquiSeq’s DNA tests for three genetic variants in horses that predispose to muscle disease. The tests are part of EquiSeq’s current DNA panel. Test results allow horse owners to better manage affected horses, and can be used to improve a breeding program to reduce the incidence of muscle disease. This is the third patent issued to EquiSeq. The same patent remains pending before patent examiners in Australia, the European Union, and the United States. A second patent is pending in the same countries. Generatio GmbH performs EquiSeq’s DNA tests for the European Union and United Kingdom, while EquiSeq performs genetic testing for horse owners in the United States and the rest of the world outside of the EU and UK.
29 Nov 2024
News
New Zealand issues patent on EquiSeq DNA tests
ALBUQUERQUE, NEW MEXICO EquiSeq announced today that New Zealand has issued a patent on EquiSeq’s DNA tests for three genetic variants in horses that predispose to muscle disease. The tests are part of EquiSeq’s current DNA panel. Test results allow horse owners to better manage affected horses, and can be used to improve a breeding program to reduce the incidence of muscle disease. This is the second patent issued to EquiSeq. The same patent remains pending before patent examiners in Australia, South Africa, the European Union, and the United States. A second patent is pending in the same countries. Generatio GmbH performs EquiSeq’s DNA tests for the European Union and United Kingdom, while EquiSeq performs genetic testing for horse owners in the United States and the rest of the world outside of the EU and UK.
15 Nov 2024
News
Canada issues patent on EquiSeq DNA tests
ALBUQUERQUE, NEW MEXICO EquiSeq announced today that Canada has issued a patent on EquiSeq’s DNA tests for three genetic variants in horses that predispose to muscle disease. The tests are part of EquiSeq’s current DNA panel. Test results allow horse owners to better manage affected horses, and can be used to improve a breeding program to reduce the incidence of muscle disease. This is the first patent issued to EquiSeq. The same patent remains pending before patent examiners in Australia, New Zealand, South Africa, the European Union, and the United States. A second patent is pending in the same countries. Generatio GmbH performs EquiSeq’s DNA tests for the European Union and United Kingdom, while EquiSeq performs genetic testing for horse owners in the United States and the rest of the world outside of the EU and UK.
29 Sep 2024
Blog
Genes First!
EquiSeq has released a major revision to the software that powers our website and database. We now present genetic test results starting with the affected gene. Here is an example of the appearance of the new horse profile pages. Figure 1. A horse profile page returning results from EquiSeq. This horse has tested positive for the K1 variant of COL6A3 (n/K1), negative for the other five variants in EquiSeq’s DNA test, and also negative for the P5 and P6 variants of DYSF, an experimental test not available commercially. What is a gene? In the context of mammalian genetics, a gene is a unit of hereditary information made of DNA. These segments of DNA encode proteins. Every mammal has two copies of every gene (except for X-linked genes in males). In a population, there are variant forms of genes, called alleles of that gene, that differ in their DNA sequence. Some alleles produce proteins that differ from the reference or wild-type sequence in minor ways that do not affect protein function. Other alleles produce proteins that are nonfunctional or have an altered function, causing a change in the development or function of the organism. What is a disease state? Disease is defined…
24 Sep 2024

Blog
Study of horse genomes explores genetic burden
A team of researchers at the University of Minnesota and the University of California, Davis, have published a landmark study of the predicted genetic burden in horses, based on the analysis of whole genome sequence data from 605 horses (1). They conclude that the genetic load in horses is 1.4 – 2.6 times that of the human population. The authors discuss the unique advantages of the study of horse to understand human phenotypes, especially those associated with athletic performance.
Horse owners have asked us to explain this paper, as it mentions the genetic variants that are in EquiSeq’s panel of DNA tests. Here we review the methods and major findings of this paper. We include background information typically absent from the primary literature in order to make the paper more accessible to non-specialists.
21 Jun 2024

Blog
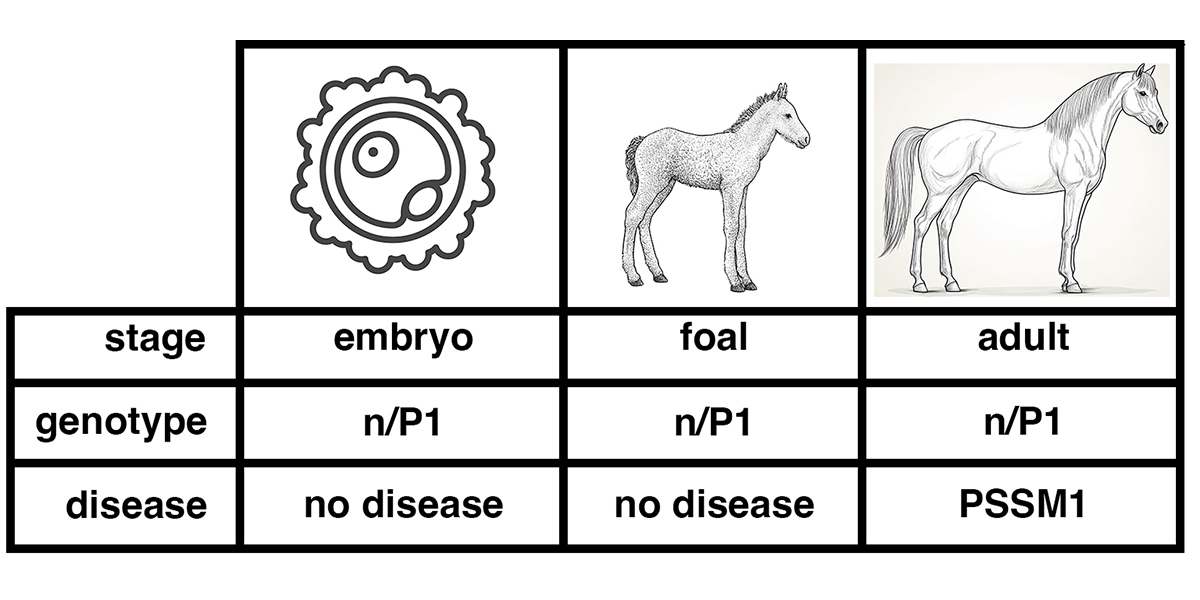
What is PSSM?
Polysaccharide Storage Myopathy (PSSM) is a form of equine exercise intolerance characterized by episodes of tying up. Horses with this condition have a particular set of clinical signs, including displays of pain, refusal to move forward, trouble standing for the farrier, and standing “parked out” as if to urinate. All of these are signs of a disease state affecting muscle. Affected horses often show enlarged granules of glycogen in muscle tissue obtained by biopsy. These enlarged glycogen granules are relatively resistant to digestion by amylase, an enzyme that completely degrades normal glycogen granules under standard conditions. Glycogen is a polysaccharide, a polymer of the simple sugar glucose, a monosaccharide. The synthesis of glycogen from glucose in muscle tissue provides a store of energy. When energy is needed, the glycogen is broken down into glucose, which can be metabolized for energy. The image above shows a) the chemical structure of glucose, b) the general structure of glycogen in which glucose units are joined in a branching structure, and c) the chemical structure of the linear polymers of glucose and the branch points. The synthesis of glycogen from glucose is highly regulated. In normal horses, glycogen is synthesized when energy needs are…
30 Jan 2024

News
Generatio Scientist Visits EquiSeq
Dr. Melissa Cox, a scientist at Generatio, visited with Chief Scientific Officer Dr. Paul Szauter at EquiSeq’s office in Albuquerque. Dr. Cox was visiting for a friend’s wedding, and made time for a two-hour discussion of scientific work at Generatio GmbH. Dr. Cox reviewed the results of a survey of horse owners exploring symptoms in over 2,000 tested horses. A first analysis of the results appeared to show a statistical association of most of the variants with particular symptoms. With additional work, this study is expected to be ready for peer review in early 2024. Drs. Cox and Szauter also discussed ongoing work at EquiSeq using cell culture, a yeast assay, and experiments in zebrafish aimed at discovering the effects of specific variants on the proteins encoded by COL6A3, MYOZ3, PYROXD1, and FLNC. The meeting was a reunion for the two scientists, who overlapped at The Jackson Laboratory years ago. When EquiSeq began marketing its tests, Dr. Cox reached out about the possibility of licensing EquiSeq’s genetic tests. The conversation moved from email to a first phone call, where the two discovered that they had both been employed at The Jackson Laboratory at the same time. Dr. Cox recalled attended…
24 Aug 2023

News
Free Testing for Standardbreds and Haflingers
Albuquerque, New Mexico A collaborative research team assembled by researchers at EquiSeq will end its offer of free testing for Standardbreds and Haflingers on March 31, 2023. The project is aimed at studying the K1 variant of COL6A3, which is present in high frequency in these breeds. The K1 variant affects the structure of collagen in muscle tissue. Collagen is part of the connective tissue that surrounds muscle fibers. Human patients with similar mutations have a myopathy called COL VI-related dystrophy. The K1 variant affects the organization of connective tissue in a cell culture assay, as reported by Chief Scientific Officer Paul Szauter at the 20th Anniversary Symposium of the Genome Sciences Department at the University of Washington in November 2022. Kirsten Dimmler, formerly a Bioinformatics Analyst at EquiSeq and now a graduate student with Dr. Molly McCue at the University of Minnesota, reported additional findings at the Plant and Animal Genomes conference in January 2023. The free testing program is open to all Standardbreds and Haflingers in the United States. Horse owners seeking testing will receive a consent form and hair sample pack by mail. To participate, send an email to contact@EquiSeq.com with the number of Standardbreds or Haflingers you…
1 Feb 2023

News
Paul Szauter Invited to Speak at Genome Sciences Symposium
Seattle, WA Paul Szauter, EquiSeq’s Chief Scientific Officer, has been invited to speak at the Genome Sciences 20th Anniversary Symposium at the University of Washington in Seattle. The Symposium will be held November 10 – 11; the schedule includes presentations by Bill Gates, Francis Collins, George Church, and Genome Sciences Chair Robert Waterston. Dr. Szauter received his PhD in Genetics from the University of Washington in 1980. Following an academic career as a Drosophila geneticist, he made a career transition to mammalian genomics and bioinformatics at The Jackson Laboratory in 2000. In 2011, he moved to New Mexico, where he worked at the University of New Mexico before entering the private sector in 2014. He founded EquiSeq in 2015. Dr. Szauter will present current research on the genetics of exercise intolerance in horses, including new results on COL6A3 and MYOZ3. The presentation, scheduled for 3:50 pm Pacific time on November 11, will be livestreamed on Zoom.
21 Oct 2022
EquiSeq is a biotech company offering
genetic testing for horses.
Resources
